È un esame diagnostico per immagini: permette di analizzare la retina e, in particolare, la macula (area centrale del tessuto retinico che ci consente di leggere, vedere i volti, ecc.) e del nervo ottico. Si tratta pertanto di un’indagine molto utile per la diagnosi ed il follow-up di diverse patologie oculari In particolare, l’oct della macula (vedi figura sopra), è in grado di fornire una serie di immagini in sezione trasversale della retina, contenenti informazioni preziose sul suo spessore, sulla sua conformazione e sul rapporto tra i vari strati che la compongono.
Come funziona?
L’ OCT si basa su una tecnica di misurazione ottica chiamata interferometria a bassa coerenza. Il principio di funzionamento è simile a quello dell’ecografia (dove però le onde sono acustiche): in pratica, sfruttando la riflessione di un fascio laser non nocivo, si riesce ad analizzare le strutture oculari ottenendo delle sezioni. . È comunque un esame più preciso di quello ecografico perché, grazie agli apparecchi di ultima generazione, consente di ottenere una risoluzione elevata (nell’ordine dei micrometri) e quindi un livello di dettaglio superiore.
Qual è la procedura?
Si eseguono delle scansioni mediante strumentazioni computerizzate, che consentono di ottenere un’immagine dettagliata della singole strutture oculari. A livello retinico, ad esempio, si possono apprezzare e analizzare i singoli strati attraverso un’analisi qualitativa e quantitativa: si rilevano con estrema precisione eventuali alterazioni, soprattutto della macula. Le fotografie retiniche scattate a diverse profondità sono chiamate “tomogrammi”.
Si tratta di un esame invasivo?
Assolutamente no. Si tratta di un esame rapido e semplice, che non necessita dell’instillazione del collirio midriatico (senza dilatazione della pupilla) né la somministrazione di mezzo di contrasto. Le scansioni OCT, come già anticipato in precedenza, possono essere utilizzate per la diagnosi e il monitoraggio di patologie che colpiscono la retina centrale (la macula), il nervo ottico e la cornea.
Come si esegue l’esame e quanto dura?
Il paziente viene fatto posizionare di fronte allo strumento ed invitato dall’operatore a fissare una mira luminosa. Le scansioni vengono eseguite piuttosto rapidamente ed infatti l’esame dura circa 15 minuti. Le immagini ottenute, possono essere analizzate, archiviate e confrontate nel tempo con quelle di esami successivi.
Per quali patologie è indispensabile effettuare l’OCT?
Optical Coherent Tomography
L’OCT viene usato più frequentemente nelle seguenti patologie:
corioretinopatia sierosa centrale (vedi maculopatia)
fori maculari (in questo caso l’OCT permette di riconoscere i diversi stadi evolutivi ed è, quindi, utile anche per la prognosi).
Ad oggi, l’OCT, è diventato un esame estremamente importante anche per quanto riguarda la diagnosi e lo studio dell’evoluzione della patologia glaucomatosa, consente infatti di misurare lo spessore dello strato delle fibre nervose retiniche (RNFL) e valutare i vari parametri della papilla ottica, ad esempio l’escavazione. Una diminuzione dello spessore delle fibre nervose retiniche e un aumento dell’escavazione papillare sono considerati segni precoci di glaucoma.
L’OCT del segmento anteriore viene invece utilizzato per lo studio dell’angolo irido-corneale, delle distrofie e degenerazioni corneali, delle neoformazioni del segmento anteriore. Consente, inoltre, di eseguire la pachimetria corneale, ovvero la misurazione dello spessore della cornea, sia centrale che periferico, dato fondamentale nella diagnosi di glaucoma, ma utile anche nei pazienti affetti da patologie corneali, quali ad esempio il cheratocono, o in quei pazienti che vogliano sottoporsi a chirurgia refrattiva.
Si può considerare esame sostitutivo alla fluorangiografia?
No. Infatti l’OCT è complementare alla fluorangiografia, soprattutto per patologie dove è importante valutare il comportamento del mezzo di contrasto nel tempo ad esempio nelle maculopatie essudative, in alcuni casi di corioretinopatia sierosa centrale o nei casi di dubbia diagnosi. Quindi completa, ma non sostituisce, l’esame obiettivo oftalmoscopico e la fluorangiografia. Da alcuni anni è disponibile anche un esame strumentale che consente di studiare i vasi senza utilizzare il mezzo di contrasto: l’angiografia OCT (abbreviata con angio-OCT).
Si può effettuare sempre?
Si può effettuare quasi sempre, tranne seguenti casi:
opacizzazione dei mezzi diottrici oculari (ad esempio cataratta avanzata), opacità massive della cornea ( èdema o leucomi), presenza di sangue o olio di silicone nella camera vitrea;
instabilità della fissazione (come il nistagmo): può rendere molto difficile il corretto posizionamento della scansione e, quindi, il confronto con un esame ripetuto in un secondo momento.
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus Leggi le condizioni generali di consultazione di questo sito
Pagina pubblicata il 17 dicembre 2008. Ultimo aggiornamento: 13 giungo 2023.
Le distrofie retiniche sono un gruppo eterogeneo di patologie oculari rare, la maggior parte delle quali ha un’origine genetica. Sono caratterizzate da un’alterazione morfofunzionale che si sviluppa a seguito di modificazioni del normale trofismo retinico. Possono colpire non solo la retina, ma anche la coroide, provocando gravi danni visivi. Ad oggi sono riconosciute clinicamente diverse forme di distrofia retinica, differenti tra di loro per età d’insorgenza, manifestazioni cliniche, gravità, rapidità evolutiva e tipo di ereditarietà.
COSA SONO LE MALATTIE RARE?
Per malattie rare s’intende un gruppo eterogeneo di patologie che colpiscono l’uomo, definite tali per la loro bassa prevalenza nella popolazione. Viene infatti considerata rara, ogni malattia che colpisce non più di 5 abitanti su 10.000.
Circa l’80% dei casi è di origine genetica, mentre per il restante 20% si parla di malattie multifattoriali, dipendenti cioè da svariate cause come ad esempio: suscettibilità individuale, fattori ambientali, fattori alimentari o interazione tra cause genetiche e ambientali.
Le malattie rare possono colpire varie fasce d’età, alcune possono manifestarsi già in fase prenatale, altre alla nascita o durante la primissima infanzia, altre ancora in età adulta. Gli elementi principali che accomunano le malattie rare sono: diagnosi difficoltosa e non sempre raggiungibile in tempi brevi, andamento cronico, rara disponibilità di trattamenti terapeutici efficaci. Lo scopo della ricerca scientifica è quindi quello di approfondire sempre di più le possibili cause delle malattie rare, in modo da riuscire a sviluppare terapie nuove ed efficaci.
QUALI SONO LE CAUSE DELLE DISTROFIE RETINICHE?
Come accennato in precedenza, le cause delle anomalie che colpiscono la retina, sono per la maggior parte da ricercarsi in mutazioni genetiche trasmissibili su base ereditaria. È sicuramente molto complicato isolare le alterazioni genetiche responsabili delle varie malattie, anche se ad oggi sono stati fatti molti passi in avanti e si è riusciti ad identificare mutazioni in più di 270 geni diversi. Il sottogruppo più frequente è quello della famiglia delle retiniti pigmentose caratterizzate da percezione luminosa ridotta e riduzione progressiva del campo visivo. Le conoscenze sui meccanismi patogenetici di queste malattie si sono notevolmente incrementate grazie alle tecniche sempre più avanzate di genetica molecolare.
Il denominatore comune di tutte le distrofie retiniche è l’estrema eterogeneità. Lo spettro di presentazione è quanto mai ampio, anche nei membri di una stessa famiglia; velocità di progressione e severità variano tantissimo, così come il quadro clinico che può differire profondamente in pazienti affetti dalla stessa patologia. L’eterogeneità riguarda anche gli aspetti genetici. Esiste una sorta di “sovrapposizione” tra quadri clinici e geni responsabili, infatti, uno stesso gene può dare origine a forme cliniche estremamente diversefra loro.
QUALI SONO I SINTOMI?
I pazienti affetti da distrofia retinica possono presentare una serie di sintomi visivi, come ad esempio:
comparsa di macchie scure nel campo visivo (scotomi);
calo del visus;
difficoltà di adattamento nel passaggio dalla luce al buio;
comparsa di metamorfopsie (percezione distorta o deformata degli oggetti, che indica alterazioni a livello della parte centrale della retina, ossia la macula);
fastidio alla luce (fotofobia);
anomala percezione dei colori;
alterazioni del campo visivo.
COME SI EFFETTUA LA DIAGNOSI?
Le distrofie retiniche possono essere diagnosticate in molti modi. Spesso l’esame diretto ed accurato del fondo oculare può già fornire informazioni rilevanti per una diagnosi precisa e affidabile, in altri casi, saranno necessari esami strumentali complementari per comprendere al meglio le caratteristiche della malattia: l’autofluorescenza e l’OCT (tomografia a coerenza ottica) permettono di eseguire un esame accurato della morfologia oculare, esami come l’elettroretinogramma (ERG), l’elettrooculogramma (EOG) e i potenziali evocati visivi (PEV), consentono di valutare/approfondire la situazione a livello funzionale. In presenza di dubbio diagnostico o per una conferma finale, si può far ricorso all’indagine genetica per rilevare la presenza del gene o dei geni mutati, responsabili dello sviluppo della malattia.
In caso di cecità retinica congenita, oltre agli esami già indicati, può essere eseguita una RMN dell’encefalo per escludere ulteriori problematiche a livello del sistema nervoso centrale.
CLASSIFICAZIONE CLINICA DELLE DISTROFIE RETINICHE
Si riconoscono diverse forme di distrofie retiniche, le principali sono le seguenti:
amaurosi congenita di Leber;
distrofia dei coni e dei bastoncelli;
distrofia ialina della retina;
distrofia vitelliforme di Best;
distrofia vitreoretinica;
malattia di Stargardt;
retinite pigmentosa;
retinite puntata albescente.
AMAUROSI CONGENITA DI LEBER?
L’amaurosi congenita di Leber (ACL) è una malattia genetica che colpisce la retina e si trasmette con modalità autosomica recessiva. La ACL provoca cecità o grave riduzione dell’ acuità visiva centrale fin dalla primissima infanzia, infatti in genere l’esordio è entro i primi sei mesi di vita. Gran parte delle persone affette dalla Leber presentano segni e sintomi caratteristici, come nistagmo, fotofobia, cecità notturna, strabismo convergente, alterazione della percezione dei colori, ipermetropia, alterazioni del campo visivo, cheratocono.
La distrofia dei coni e dei bastoncelli è una grave forma di malattia retinica che colpisce i fotorecettori, le cellule nervose dell’occhio che trasformano il segnale luminoso in segnale elettrico per generare poi la risposta visiva. Ci sono due tipi di fotorecettori, i coni e i bastoncelli, i primi sono concentrati nella parte centrale della retina e sono deputati alla visione dei colori e alla visione distinta, i secondi si concentrano invece nella zona periferica della retina e sono utilizzati per la visione al buio. La distrofia dei coni e dei bastoncelli, compare nei primi sei mesi di vita, può portare a cecità o ipovisione ed è caratterizzata dalla contestuale presenza di nistagmo. Di solito la patologia non si associa a malformazioni o disfunzioni a carico di altri organi e apparati. A livello sintomatologico i piccoli pazienti presentano una progressiva diminuzione della visione centrale e della capacità di distinguere i colori, oltre ad una spiccata fotofobia. Ad oggi, sono stati identificati una decina di geni associati alla distrofia dei coni e alle sue varianti, quello più frequentemente coinvolto è ABCA4, che causa anche la malattia di Stargardt, ed ha una trasmissione di tipo autosomico-recessivo (per manifestare i sintomi occorre ereditare la mutazione genetica da ciascuno dei genitori, entrambi portatori sani).
Purtroppo, ad oggi, non esiste alcuna terapia risolutiva, per attenuare i disturbi visivi è possibile utilizzare lenti dotate di filtri per proteggersi dalla luce. Gli ausili per ipovedenti possono essere utili se l’acuità visiva è piuttosto ridotta.
DISTROFIA IALINA DELLA RETINA
La distrofia ialina della retina è caratterizzata dalla perdita graduale della vista, cecità notturna e presenza di segni oculari caratteristici quali alterazioni del corpo vitreo, retinoschisi maculare, atrofia corioretinica, sviluppo precoce della cataratta, distacco di retina. La malattia colpisce in ugual misura sia maschi che femmine e si manifesta clinicamente entro i primi venti anni d’età, ma l’ERG può risultare estinto o fortemente alterato già entri i primi anni di vita. La trasmissione è autosomica recessiva. Non è disponibile al momento una terapia, può essere indicato eseguire dei trattamenti laser retinici per prevenire il distacco di retina.
DISTROFIA VITELLIFORME DI BEST
La distrofia vitelliforme di Best è una patologia ereditaria della retina. Viene trasmessa in forma autosomica dominante (un genitore trasmette il difetto genetico al figlio o alla figlia). La malattia è causata da una mutazione di un gene (chiamato VMD2, localizzato sul cromosoma 11q13), che nella retina regola il trasporto di determinate sostanze (acidi grassi polinsaturi) e comporta l’accumulo di un materiale di scarto biologico (lipofuscina) in uno strato della retina chiamato epitelio pigmentato retinico. I sintomi consistono nella riduzione della vista generalmente in forma lieve, con una progressione lenta. I pazienti riferiscono disturbi maggiori nelle visione da vicino, a cui si possono accompagnare la distorsione dell’immagine e gli scotomi centrali (macchie nere nel campo visivo).
Per un approfondimento leggi anche la scheda sulla malattia di Best.
MALATTIA DI STARGARDT
La malattia (o maculopatia) di Stargardt è una patologia ereditaria della retina che si manifesta generalmente prima dei vent’anni. Il più delle volte viene trasmessa in forma autosomica recessiva (entrambi i genitori presentano il difetto genetico pur potendo essere portatori sani), ma sono stati descritti anche casi di forme autosomiche dominanti (un solo genitore trasmette il difetto del DNA). La malattia è provocata da una mutazione di un gene (ABCA4), che comporta l’accumulo di materiale di scarto (simile alla lipofuscina) nella retina (in uno strato esterno chiamato epitelio pigmentato). Questo materiale è originato dalla degradazione di sostanze presenti nei coni e nei bastoncelli (fotorecettori retinici). I sintomi, consistono soprattutto nella riduzione della visione centrale (spesso in forma grave) che può iniziare durante l’adolescenza o anche nell’infanzia. Inoltre, chi ne è affetto può lamentare disturbi nella percezione dei colori (discromatopsia), scotomi centrali (macchie nere nel campo visivo) e fotofobia (intolleranza alla luce).
Si tratta di una patologia rara di tipo ereditario, caratterizzata da una degenerazione progressiva della retina in entrambi gli occhi. Provoca la perdita graduale della visione notturna e del campo visivo periferico, ma agli ultimi stadi si può verificare anche una perdita della visione centrale. I principali sintomi che possono indurre il medico a sospettare di trovarsi di fronte ad un caso di retinite pigmentosa sono essenzialmente due: cecità crepuscolare e notturna e restringimento del campo visivo (visione tubulare).
La retinite puntata albescente è una forma atipica e progressiva di retinite pigmentosa, con modalità di trasmissione autosomica recessiva, caratterizzata dalla presenza di chiazzette retiniche biancastre. Tali chiazzette, sparse su tutta la retina, possono precedere o coesistere con la pigmentazione tipica della retinite pigmentosa. I sintomi caratteristici della malattia sono la cecità notturna e il restringimento progressivo del campo visivo, l’ ERG può risultare fortemente alterato o estinto. Purtroppo non esistono ad oggi trattamenti terapeutici efficaci.
E’ POSSIBILE CURARE LE DISTROFIE RETINICHE?
Attualmente non ci sono delle terapie per la cura delle distrofie retiniche. Sono molte però le strade di ricerca aperte, i filoni più promettenti sono la terapia genica, il ricorso alle cellule staminali, il trapianto di retina, l’occhio bionico.
Pagina pubblicata nel 2023. Ultimo aggiornamento: 23 giugno 2023.
Ultima revisione scientifica:23 giugno 2023.
Contatta l’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Il Numero Verde di consultazione oculistica è attivo dal lunedì al venerdì dalle 10 alle 13. Scrivi nel Forum: un medico oculista ti risponderà gratuitamente.
Si tratta di una patologia che colpisce il cristallino, la lente contenuta all’interno del nostro occhio, rendendolo opaco già alla nascita o entro i primi 3 mesi di vita. Le opacità possono essere di dimensioni limitate ed in tal caso interferiscono poco o affatto sulla visione, oppure possono presentarsi come estese e dense, andando ad incidere molto sul visus. La cataratta congenita può rimanere stabile o evolversi nel corso degli anni, può inoltre interessare un solo occhio (cataratta congenita monolaterale) o entrambi (cataratta congenita bilaterale).
Quant’è frequente?
Ancora oggi rappresenta una delle cause più frequenti di cecità nell’infanzia (10-15%). In circa i 2/3 dei casi sono coinvolti entrambi gli occhi (cataratta bilaterale congenita).
Quali sono le cause?
Stabilire la causa dell’opacità del cristallino è fondamentale per un completo inquadramento clinico, però diversi sono i fattori che possono essere responsabili di una cataratta congenita:
causa idiopatica (non dovuta a cause esterne note ovvero senza causa apparente);
fattori genetici: la trasmissione ereditaria più frequente è quella autosomica dominante. Talvolta l’eziologia può essere un’alterazione cromosomica, come la trisomia 21 o sindrome di Down, la trisomia 13, la trisomia 18, la sindrome di Turner;
esposizione della madre durante la gravidanza a trattamenti con raggi X, con particolare rischio se praticata durante i primi 3 mesi di gravidanza;
infezioni contratte dalla madre durante la gravidanza, come rosolia, parotite e varicella;
farmaci assunti dalla madre durante la gravidanza, soprattutto corticosteroidi e alcuni antibiotici (specialmente i sulfamidici);
alterazioni metaboliche dovute – nelle madri incinte – a diabete, ipoparatiroidismo o gravi carenze alimentari e – nel feto – alla galattosemia (malfunzionamento di un enzima capace di metabolizzare il galattosio);
anomalie dell’iride, quali aniridia(assenza congenita dell’iride, completa o quasi, in entrambi gli occhi), alterazioni delle strutture anatomiche anteriori dell’occhio, persistenza del vitreo primitivo iperplastico (di solito l’occhio colpito è più piccolo della norma e ha problemi funzionali), microftalmo (malattia ereditaria per cui l’occhio è, anche in questo caso, più piccolo del normale) e retinopatia del prematuro (ROP );
la prematurità e la sofferenza feto-neonatale;
malattie sistemiche associate del neonato (artrite reumatoide, sindrome di Marfan, sindrome di Weill-Marchesani e malformazioni cranio-facciali).
Di che tipo di opacità si tratta?
Come già anticipato in precedenza le opacità del cristallino possono essere singole o multiple, con dimensioni più o meno estese e di diversa densità. In base a come si presenta l’opacità del cristallino possiamo classificare la cataratta congenita in:
polare anteriore;
polare posteriore;
pulverulenta;
centrale pulverulenta;
zonulare;
totale.
Come si esegue la diagnosi di cataratta congenita?
Se si sospetta la presenza di una cataratta congenita, si può fare il testdel riflesso rosso. Si tratta di un esame piuttosto veloce e non invasivo, da eseguire in una stanza oscurata, per consentire un buon allargamento delle pupille del neonato. Il medico esaminatore proietta una luce in entrambi gli occhi del piccolo paziente da una distanza di circa 45 cm, dopodiché osserva come si presenta il riflesso rosso. In condizioni di normalità tale riflesso deve essere presente e simmetrico in ambedue gli occhi. Se si evidenziano invece macchie nere nel riflesso, un riflesso marcatamente diminuito, la presenza di un riflesso bianco o l’asimmetria dei riflessi, vuol dire che siamo in presenza di un’anomalia e che è necessario un approfondimento con visita completa per una conferma diagnostica.
Sintomi della cataratta congenita
Le opacità estese e molto dense del cristallino determinano un’ambliopia più o meno grave (occhio pigro), in quanto impediscono il normale sviluppo funzionale dell’apparato visivo, che avviene proprio nei primi mesi di vita grazie alla stimolazione delle vie ottiche ad opera delle immagini provenienti dall’esterno. Generalmente sono gli stessi genitori a segnalare la presenza di un riflesso pupillare biancastro (leucocoria) o l’assenza del classico riflesso rosso in fotografia. Se la cataratta è monolaterale, oltre all’ambliopia si sviluppa generalmente strabismo. In presenza di cataratta bilaterale evoluta un segno tipico può essere il nistagmo che insorge intorno ai 3 mesi di età. Risulta fondamentale, quindi, una diagnosi precoce: prima si interverrà e più possibilità di recupero funzionale visivo ci saranno.
Si può curare?
Sì, generalmente è trattabile con un’operazione chirurgica. Ovviamente alcune cataratte infantili che non interferiscono sulla capacità visiva, in quanto piccole o poco dense, non richiedono la chirurgia.
Qual è la terapia più adatta?
Se la cataratta ostacola in maniera grave lo sviluppo della funzione visiva è fondamentale intervenire chirurgicamente il più presto possibile, asportando il cristallino opacizzato. Si consiglia di effettuare l’intervento entro i primi 3 mesi di vita. In prospettiva si potrebbero utilizzare persino cellule staminali del cristallino stesso per “rigenerarlo“ (vedi Nature); tuttavia quest’approccio, al momento in cui scriviamo è da considerarsi meramente sperimentale e attualmente privo di applicazioni cliniche.
Il cristallino asportato viene sostituito con un cristallino artificiale come nella cataratta senile?
Generalmente si preferisce inserire la lentina artificiale se il bambino ha più di 18 mesi. Fino ad allora si deve proseguire con un trattamento riabilitativo, in quanto l’occhio senza cristallino vede ovviamente male; a questo scopo si utilizzano occhiali adeguati o, se la cataratta riguarda un solo occhio, una lente a contatto morbida. La terapia chirurgica rappresenta, quindi, solo il primo passo di un lungo percorso terapeutico
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus Leggi le condizioni generali di consultazione di questo sito
Pagina pubblicata il 10 dicembre 2010. Ultimo aggiornamento:18 aprile 2023
Ultima revisione scientifica: 18 aprile 2023
Contatta l’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Il Numero Verde di consultazione oculistica è attivo dal lunedì al venerdì dalle 10 alle 13. Scrivi nel Forum: un medico oculista ti risponderà gratuitamente.
“Oftalmologia Sociale” è una rivista di sanità pubblica, la pubblicazione trimestrale dell’Agenzia Internazionale per la prevenzione della Cecità-IAPB Italia onlus.
“Oftalmologia Sociale” è una rivista di sanità pubblica, la pubblicazione trimestrale dell’Agenzia Internazionale per la prevenzione della Cecità-IAPB Italia onlus.
“Oftalmologia Sociale” è una rivista di sanità pubblica, la pubblicazione trimestrale dell’Agenzia Internazionale per la prevenzione della Cecità-IAPB Italia onlus.
“Oftalmologia Sociale” è una rivista di sanità pubblica, la pubblicazione trimestrale dell’Agenzia Internazionale per la prevenzione della Cecità-IAPB Italia onlus.
Contatta l’Agenzia internazionale per la prevenzione della cecità-IAPB Italia ETS
Il Numero Verde di consultazione oculistica è attivo dal lunedì al venerdì dalle 10 alle 13. Scrivi nel Forum: un medico oculista ti risponderà gratuitamente.
Consiste in una lesione della parte esterna della cornea con perdita parziale di tessuto superficiale (epitelio).
Quali sono le cause?
Le abrasioni più comuni sono quelle traumatiche causate da diversi fattori: corpi estranei, unghiate, ditate, rottura di lenti a contatto, foglie d’albero o di piante , esposizione senza protezione ai raggi ultravioletti (UV), ecc. Possono essere coinvolti gli strati più superficiali della cornea o, nei casi più complicati, possono essere interessati gli strati profondi. Di solito è necessario eseguire una visita in urgenza, rivolgendosi al proprio oculista o recandosi presso il pronto soccorso oculistico più vicino.
Quali sono i sintomi provocati dal trauma?
In presenza di un’abrasione corneale i sintomi più comuni sono: sensazione di corpo estraneo, dolore, fotofobia, arrossamento oculare e lacrimazione abbondante. Se però è colpita la zona centrale della cornea ci sarà anche un’alterazione più o meno importante del visus perché viene coinvolta la zona ottica che si trova in corrispondenza della pupilla (il visus può presentarsi sfocato/appannato, come se si stesse guardando attraverso un vetro sporco).
Come si esegue la diagnosi?
Lo specialista, oltre ad osservare la superficie anteriore dell’occhio alla lampada a fessura, aiutandosi con un colorante specifico (fluoresceina) – con cui si evidenziano anche le più piccole abrasioni corneali –, aprirà e rovescerà la palpebra superiore (eversione palpebrale) per escludere la presenza di eventuali corpi estranei. Se la lesione coinvolge gli strati più profondi della cornea e si sospetta che il corpo estraneo sia penetrato all’interno del bulbo oculare, bisognerà eseguire una radiografia per individuarlo .
Si può prevenire?
Sì, in particolare con l’uso di occhiali protettivi sui luoghi di lavoro a rischio e durante le attività sportive che implicano un contatto diretto.
Sì: se non è profonda la lesione della cornea si può rimarginare nell’arco di 48-72 ore. La terapia varia a seconda dell’entità della lesione. Generalmente l’abrasione corneale viene curata con l’applicazione di una pomata antibiotica, e bendaggio dell’occhio per almeno 2-3 giorni consecutivi; bisogna, infatti, dare il tempo al tessuto corneale di riformarsi. Nei casi di forte dolore è consigliabile l’uso di antinfiammatori non steroidei per bocca.
Per le abrasioni indotte da agenti penetranti bisogna eseguire interventi chirurgici specifici per ottenere una perfetta chiusura della ferita e ristabilire così l’integrità delle strutture anatomiche lese. Invece per le abrasioni causate da sostanze chimiche è importante lavare rapidamente l’occhio, rimuovendo così l’agente ustionante e poi provvedere immediatamente a farsi visitare.
Cosa è necessario fare se è entrata una scheggia nell’occhio?
Generalmente lo specialista la rimuove a livello ambulatoriale, dopo aver anestetizzato l’occhio instillando un collirio, tranne alcuni casi in cui è necessario un intervento chirurgico (se il corpo estraneo è penetrato in profondità). In quest’ultimo caso si possono determinare alterazioni delle strutture anatomiche oculari. Infatti, la velocità e la forma del corpo estraneo penetrato nel bulbo può causare un danneggiamento del cristallino – con conseguente cataratta traumatica – e danni alla retina, con rotture o distacco traumatico.
Fondamentale è la rimozione della scheggia: la persistenza di eventuali residui (come ad esempio della ruggine nel caso di corpi estranei di ferro) può creare ulteriori danni e la non completa guarigione della lesione. È necessario poi intraprendere una terapia con pomata antibiotica e usare un collirio per dilatare la pupilla, oltre a un bendaggio volto a immobilizzare la palpebra dell’occhio interessato.
Se il danno è dovuto a una rottura delle lenti a contatto che bisogna fare?
Dopo l’intervento dell’oculista, che provvederà all’asportazione del frammento della lente a contatto, si attuerà una terapia antibiotica locale. Bisognerà attendere poi la completa riepitelizzazione corneale (ricostituzione degli strati della superficie oculare) per poter indossare nuovamente le lenti a contatto. È opportuno astenersi però da un loro impiego per almeno due settimane dopo la cicatrizzazione della lesione.
Se si ha il sospetto di avere un frammento di lente a contatto trattenuto nell’occhio (nella maggior parte dei casi si posiziona sotto la palpebra superiore), ma non si riesce a vederlo e a rimuoverlo, non è consigliabile eseguire manovre traumatiche e/o vigorose, strofinando gli occhi, provando a rigirare la palpebra ecc, perché si rischia di complicare la situazione, causando infiammazione dell’occhio e magari provocando involontariamente delle lesioni sulla cornea. In questi casi è sempre bene rivolgersi all’oculista.
Cosa fare se l’abrasione è causata dai raggi ultravioletti (UV)?
L’abrasione avviene solo in casi gravi, come per un mancato uso della maschera protettiva (dotata di filtri speciali) durante lavori di saldatura o degli occhiali protettivi durante l’esposizione a lampade abbronzanti. La terapia da attuare è di solito a base di pomata antibiotica locale, seguita da bendaggio per almeno 2-3 giorni. In caso di lesione bilaterale (entrambi gli occhi coinvolti) si benderà l’occhio più gravemente colpito. In caso di dolore si consiglia l’assunzione di farmaci antinfiammatori non steroidei per bocca, sempre dopo aver consultato un medico.
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus Leggi le condizioni generali di consultazione di questo sito
Pagina pubblicata il 1 settembre 2010. Ultimo aggiornamento: 06 marzo 2023.
Ultima revisione scientifica: 06 marzo 2023.
Contatta l’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Il Numero Verde di consultazione oculistica è attivo dal lunedì al venerdì dalle 10 alle 13. Scrivi nel Forum: un medico oculista ti risponderà gratuitamente.
Il significato del termine xeroftalmia è occhio secco, dal greco xero “secco” e ophtalmós “occhio”. Si tratta, infatti, di una grave patologia dell’occhio, caratterizzata dalla presenza di secchezza oculare, causata dalla carenza di vitamina A. Può degenerare in ulcerazione della cornea (cheratomalacia).
Diffusione
La xeroftalmia risulta essere la maggiore causa di cecità infantile nei Paesi in via di sviluppo. Si calcola che:
250 milioni di bambini in età prescolare soffrono di carenza di vitamina A;
ogni anno 350.000 bambini diventano ciechi (complessivamente, secondo l’OMS, sono 1,4 milioni i piccoli non vedenti per diverse cause);
annualmente muoiono 2 milioni di bambini per la carenza di vitamina A (ipovitaminosi).
Aspetti Clinici
La xeroftalmia si sviluppa a seguito della perdita di cellule della congiuntiva (caliciformi e mucosecernenti) e a un’alterazione delle cellule epiteliali congiuntivali (metaplasia squamosa). Tali alterazioni causano la cheratinizzazione dell’epitelio congiuntivale, che conferisce a questo tessuto un aspetto secco: è la “xerosi congiuntivale”. Quest’ultima si manifesta a livello della congiuntiva bulbare: le caratteristiche tipiche includono la mancanza di umidità della mucosa, un suo ispessimento, un raggrinzimento e una perdita della trasparenza. A tale quadro clinico può associarsi la presenza di piccole placche grigiastre di aspetto schiumoso (più frequentemente localizzate a livello del limbus corneale, temporalmente e bilateralmente), dette macchie di Bitot. L’instabilità del film lacrimale causa un aspetto ruvido e opaco della cornea (xerosi corneale). La secchezza corneale e congiuntivale possono portare a difetti dell’epitelio corneale fino ad arrivare all’ulcerazione della stessa, con conseguente perdita della vista nel 50% dei pazienti non trattati.
Sintomi
I pazienti affetti da xeroftalmia presentano svariati sintomi, quelli più comuni sono:
emeralopia (cecità notturna);
bruciore oculare;
iperemia congiuntivale;
sensazione di sabbia nell’occhio;
fastidio alla luce (fotofobia);
secchezza oculare.
Diagnosi
L’apporto di vitamina A dovrebbe essere adeguato. Il livello plasmatico di vitamina A è l’elemento principale per la diagnosi [1]. La citologia ad impressione congiuntivale è una tecnica utile per identificare la xeroftalmia. I metodi di raccolta, fissazione e colorazione del campione sono adatti ad essere usati nei paesi in via di sviluppo, a differenza dell’altra metodica (misurazione dei livelli sierici della vitamina). Può essere utile anche eseguire il test di Schirmer, per una valutazione della quantità di lacrime prodotte. Invece le macchie di Bitot non possono essere un’adeguata spia di uno stato d’ipovitaminosi A: possono essere infatti legate ad uno stato di malnutrizione generalizzata.In alcuni casi la xeroftalmia può essere associata a xerostomia (bocca secca), in tali pazienti è consigliabile far eseguire delle indagini specifiche che escludano la presenza di una patologia autoimmune (come ad esempio la sindrome di Sjögren). Entrambi le problematiche (xeroftalmia e xerostomia) possono essere presenti anche nel lupus eritematoso sistemico, l’artrite reumatoide e la fibromialgia.
Trattamento
L’integrazione di vitamina A dà di solito buoni risultati nella regressione della cecità notturna, della xerosi congiuntivale e delle macchie di Bitot, ma è meno efficace nel trattamento delle complicanze corneali. La profilassi è sempre preferibile alla terapia: può essere somministrata oralmente o mediante iniezione intramuscolare.Oltre all’assunzione di vitamina A, è importante mantenere la superficie oculare sempre ben idratata, attraverso l’instillazione ripetuta più volte al giorno di colliri lubrificanti, da associare eventualmente ad una somministrazione in gel (sempre con prodotti lubrificanti) la sera prima di andare a dormire. Per alleviare i disturbi causati dalla secchezza oculare è, inoltre, preferibile non esporsi al vento e a fonti di calore eccessivo ed evitare di soggiornare in ambienti con aria troppo secca. Con pochissime risorse si potrebbe ridurre la mortalità dei bambini del 34% nelle aree con questa deficienza vitaminica. Il programma Vision 2020 [2] puntava ad identificare le aree a maggior rischio e ad incoraggiare misure preventive attraverso immunizzazioni, educazione a una corretta alimentazione e all’assunzione di complementi di Vitamina A.
[1] “gli indicatori clinici e funzionali correlati con la salute oculare e i biomarcatori biochimici della vitamina A (ovvero il retinolo serico, l’RBP, il retinolo derivante dall’allattamento al seno, i test di dosaggio, il metodo della diluizione isotopica e gli esteri serici del retinolo). Tali biomarcatori sono quindi correlati con le concentrazioni di vitamina A nel fegato, che di solito sono considerati il gold standard per valutare i livelli di vitamina A”, (cit. da Tanumihardjo SA, Russell RM, Stephensen CB, Gannon BM, Craft NE, Haskell MJ, Lietz G, Schulze K, Raiten DJ, “Biomarkers of Nutrition for Development (BOND)-Vitamin A Review”, J Nutr. 2016 Sep;146(9):1816S-48S. doi: 10.3945/jn.115.229708. Epub 2016 Aug 10)
Pagina pubblicata il 27 aprile 2007. Ultimo aggiornamento: 27 gennaio 2023.
Ultima revisione scientifica: 27 gennaio 2023.
Contatta l’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Il Numero Verde di consultazione oculistica è attivo dal lunedì al venerdì dalle 10 alle 13. Scrivi nel Forum: un medico oculista ti risponderà gratuitamente.
Degenerazione maculare legata all’età (AMD o DMLE)
Cos’è?
Con l’espressione “degenerazione maculare” si indica una malattia che colpisce la zona centrale della retina (la macula), provocando tutta una serie di alterazioni anatomiche e funzionali, in grado di portare ad una perdita della visione centrale. La patologia viene spesso indicata come degenerazione maculare legata all’età (AMD o DMLE), perché si manifesta principalmente in soggetti con età superiore ai 60 anni.
Cosa provoca?
È causa di un’importante e irreversibile riduzione della funzionalità visiva centrale. Il fenomeno correlato più comune è il processo d’invecchiamento dell’occhio: la macula, contenente numerosi fotorecettori (vi sono concentrati i coni), si altera sino a perdere le sue normali caratteristiche. Ciò è dovuto alla morte delle cellule retiniche, che può essere lenta e progressiva oppure più rapida e drammatica.
Quant’è diffusa?
La degenerazione maculare legata all’età (AMD o DMLE) è attualmente considerata la prima causa di cecità legale nei Paesi di maggior benessere e la terza in assoluto. Indicativamente il 5% della cecità mondiale è attribuibile all’AMD, una percentuale che sale però al 41% nei Paesi benestanti.
i prevede che, con il passare degli anni, sempre più persone saranno colpite dalla malattia a causa dell’aumento dell’invecchiamento demografico mondiale (soprattutto nei Paesi più industrializzati).
L’incidenza dell’AMD è rara prima dei 55 anni, ma aumenta soprattutto dopo i 75. La forma più grave della malattia, detta “umida”, è meno frequente e a più rapida evoluzione ma attualmente è l’unica considerata trattabile.
Quali sono i suoi sintomi?
I sintomi iniziali della degenerazione maculare può essere una distorsione delle immagini nella parte centrale del campo visivo (dove si punta lo sguardo). Altri sintomi piuttosto comuni sono: diminuzione dell’acuità visiva, difficoltà nella lettura e nello svolgimento di attività a distanza ravvicinata (in cui è richiesta la visione dei piccoli dettagli), necessità di utilizzare una fonte di luce sempre più intensa per vedere da vicino, perdita della brillantezza dei colori, difficoltà a riconoscere i volti delle persone, difficoltà nell’adattamento al buio. La degenerazione maculare comporta dunque una severa penalizzazione visiva, ma è bene sottolineare che essa (anche nei casi più gravi), non provoca la cecità totale, in quanto la visione paracentrale e quella laterale vengono solitamente conservate. Comunque si tratta di una patologia fortemente invalidante, che può avere anche gravi ripercussioni sul piano psicologico.
CAUSE
L’eziologia dell’AMD non è stata tuttora dimostrata, ma sono stati evidenziati numerosi fattori di rischio associati alla sua comparsa, quali:
età superiore ai 55 anni;
sesso (le donne sono maggiormente predisposte a sviluppare la malattia);
fumo di sigaretta;
abuso di alcol;
diabete mellito;
vita sedentaria;
dieta povera di vitamine e acidi grassi (in particolare omega-3);
ipertensione arteriosa;
disturbi della coagulazione;
esposizione prolungata e ripetuta a sorgenti di luce molto intense.
Inoltre è ormai acclarata la familiarità come principale fattore di rischio nello sviluppo della malattia da parte di soggetti con parenti di primo grado che ne sono affetti (l’origine è infatti genetica). [1]
Numerosi sono i fattori genetici che sono stati associati a un incremento del rischio di sviluppare la maculopatia. Tra questi vi sono soprattutto i geni CFH e ARMS2: in particolare la variante del gene CFH (chiamata rs1061170) è stata associata a un aumento di almeno cinque volte del rischio di ammalarsi di AMD.
Tra l’altro è possibile effettuare un test genetico mediante tampone orale per conoscere il rischio di ammalarsi di AMD, ma la sua affidabilità non è ancora molto alta [2]. Uno studio pubblicato a novembre del 2012 individua, inoltre, un meccanismo genetico che, provocando l’aumento dell’espressione di una proteina nella retina (IL17RC), promuoverebbe l’infiammazione della macula e l’attacco alle sue cellule (che di conseguenza muoiono), da parte del proprio sistema immunitario.
CLASSIFICAZIONE
Esistono due forme di degenerazione maculare legata all’età (detta anche degenerazione maculare senile), entrambe associate ad alterazioni del microcircolo capillare, tipiche dell’età avanzata: la forma secca (o atrofica) e quella umida (o essudativa); queste andrebbero considerate come due patologie distinte, poiché le loro prognosi ed eventuali terapie sono del tutto diverse.
La forma secca o atrofica(85-90% dei casi) è caratterizzata da un assottigliamento progressivo della retina centrale, che risulta scarsamente nutrita dai capillari (poco efficienti) e, di conseguenza, si atrofizza (muoiono le cellule nervose fotosensibili), determinando la formazione di una cicatrice in sede maculare con un aspetto detto a “carta geografica” (areolare).
L’altra forma di degenerazione maculare, quella più grave e a più rapida evoluzione, è detta umida o essudativa(10-15% dei casi): è complicata dalla formazione di nuovi capillari con una parete molto fragile. Questi vasi sono permeabili al plasma (la parte liquida del sangue) e possono dare origine, quindi, a distacchi sierosi dell’epitelio pigmentato retinico e, nei casi più avanzati, si possono rompere facilmente, provocando un’emorragia retinica. I ripetuti episodi emorragici e di riparazione tissutale sono responsabili della formazione di una cicatrice centrale più o meno esuberante.
Entrambe le forme di degenerazione maculare si accompagnano, a livello maculare, alle drusen, ossia a corpi colloidi: si tratta di depositi di “scarto” di forma irregolarmente rotondeggiante, situati sotto la retina (depositi subepiteliali piccoli e polimorfi). Se ne possono distinguere essenzialmente due tipi: hard drusen (meno gravi) e soft drusen (potenzialmente più nocive per la vista).
La cosiddetta fase delle drusen è generalmente priva di sintomi e solitamente non dà origine a riduzione dell’acutezza visiva. A volte si può presentare tuttavia una distorsione centrale delle immagini, principalmente delle linee rette (metamorfopsie).
Secondo uno studio pubblicato su Jama Ophthalmology il 2 aprile 2015, l’età senile e la mutazione di due alleli (CFH e ARMS2) sono i due principali fattori di rischio associati allo sviluppo di accumuli proteici. La copresenza di drusen medie e di anomalie nell’epitelio pigmentato retinico è segno di un rischio maggiore di progressione dell’AMD rispetto alla sola presenza delle drusen di medie dimensioni.
DIAGNOSI
La diagnosi di degenerazione maculare può essere fatta dall’oculista a seguito di una visita completa. In particolare lo specialista dovrà: raccogliere l’anamnesi del paziente (in modo da rilevare la presenza di eventuale familiarità per la malattia e risalire all’epoca d’insorgenza dei primi disturbi visivi); misurare l’acuità visiva (sia da lontano che da vicino); eseguire l’esame del fondo oculare, che consiste nell’osservare la retina centrale (macula) e quella periferica, tramite l’utilizzo di uno strumento detto oftalmoscopio e apposite lenti (dopo aver dilatato le pupille). Oftalmoscopicamente le drusen appaiono come piccoli depositi di colore giallastro.
Un esame molto facile da eseguire ed utilissimo per monitorare nel tempo l’evoluzione della patologia è il reticolo di Amsler (una griglia a quadretti con un punto centrale), che consente di riconoscere distorsioni o zone cieche centrali.Uno dei sintomi dellamalattia è, infatti, una distorsione delle linee rette (righe di un quaderno, linee formate dalle mattonelle del pavimento) in prossimità del centro del campo visivo. In presenza di tali alterazioni visive, è importante sottoporsi al più presto ad un controllo medico oculistico per una diagnosi precisa.
In alcuni casi, per meglio inquadrare la situazione clinica, l’oculista può richiedere anche degli esami diagnostici strumentali, quali: l’OCT (tomografia a coerenza ottica), l’angio-OCT, l’angiografia con fluoresceina (FAG) e l’angiografia al verde di indocianina (ICG). L’OCT permette di studiare le alterazioni ultrastrutturali retiniche nelle fasi precoci della malattia, visualizzando e misurando lo spessore della retina e dei singoli strati retinici; l’angio-OCT ,consente di studiare i vasi retinici senza utilizzare il mezzo di contrasto; l’angiografia con fluoresceina e quella al verde di indocianina, consentono di ottenere immagini molto dettagliate della circolazione sanguigna, sia della retina che della coroide, dopo aver iniettato in vena un mezzo di contrasto.
Tali indagini sono utili allo specialista per fare la diagnosi e studiare l’evoluzione della malattia nel tempo, oltre a essere una guida preziosa per un eventuale trattamento.
Trattamenti
A seconda che si tratti della forma secca oppure di quella umida l’approccio è differente. Le forme secche sono considerate ancora oggi incurabili; tuttavia potrebbe essere possibile, una volta diagnosticata, rallentarne almeno in parte l’evoluzione (ad esempio mediante un corretto stile di vita che va da esercizi fisici regolari a una dieta variata), anche se la questione resta scientificamente controversa. Inoltre esistono alcuni casi in cui le forme secche evolvono in quelle umide.
Alcuni ricorrono a integratori alimentari a base di sostanze antiossidanti, che potrebbero aiutare a combattere la formazione dei radicali liberi e l’ischemia del tessuto retinico maculare (ossia la sua morte dovuta alla riduzione o all’arresto dell’apporto di sangue alla retina). Quelli più comunemente utilizzati sono la luteina, le vitamine A ed E, i sali minerali (quali lo zinco, il rame e il selenio) e gli antiossidanti vegetali (quali la zeaxantina e l’astaxantina).
La terapia fotodinamica (PDT)
La forma umida (essudativa) è stata trattata, soprattutto in passato (oggi si predilige l’uso delle iniezioni intravitreali con farmaci anti-vegf), con la terapia fotodinamica, attuata mediante un tipo particolare di laser, previa iniezione endovenosa di una sostanza chiamata verteporfina. Tale trattamento laser, consente l’occlusione selettiva dei nuovi vasi (crea dei trombi che chiudono i capillari nocivi), cercando di bloccare l’evoluzione della malattia. Il trattamento può essere ripetuto nel tempo se la malattia dovesse ripresentarsi a distanza di mesi (recidiva).
Le iniezioni intravitreali
Attualmente il trattamento di prima scelta per la maculopatia essudativa è rappresentato dalle iniezioni intravitreali di farmaci anti-VEGF. Si tratta di sostanze che agiscono inibendo la proliferazione di nuovi vasi sanguigni della retina (azione antiangiogenica), che provocano la comparsa di membrane sottoretiniche e di sanguinamenti. Questi farmaci, pegaptanib sodico, bevacizumab[3], ranibizumab [4], aflibercept, e brolucizumab (sono i nomi dei principi attivi), permettono solitamente di ottenere dei buoni risultati nella cura delle degenerazioni maculari essudative, cercando di rallentare/bloccare la malattia e mirando ad ottenere una stabilizzazione del visus del paziente.
La loro somministrazione deve essere effettuata in ambiente sterile e affinché il trattamento possa essere efficace va ripetuto per alcuni mesi. Se, invece, non dovessero esserci benefici o le corrette indicazioni, ovviamente la somministrazione delle iniezioni dovrà essere sospesa.
Ricerche scientifiche relative ai primi farmaci anti-VEGF utilizzati per la maculopatia essudativa
In concomitanza all’utilizzo dei primi farmaci anti-vegf somministrati per via intravitreale, sono state condotte una serie di ricerche scientifiche per verificarne l’efficacia e la sicurezza. In particolare, una ricerca pubblicata sul New England Journal of Medicine [5] nel 2011, è giunta alla conclusione che, a distanza di un anno dall’inizio del trattamento con iniezioni intravitreali, le due molecole generalmente utilizzate (soprattutto all’inizio) contro l’AMD umida (bevacizumab e ranibizumab) avevano i medesimi effetti sull’acuità visiva. “Test clinici – puntualizzano i ricercatori dello studio chiamato CATT (Comparison of Age-related Macular Degeneration Treatments Trials) – hanno stabilito l’efficacia del ranibizumab per il trattamento della degenerazione maculare legata all’età (AMD) neovascolare. Inoltre il bevacizumab viene usato off-label (ossia esulando dalle indicazioni del foglietto illustrativo, ndr) per trattare l’AMD, nonostante l’assenza di analoghi dati a supporto”. In Italia può essere utilizzato come off-label ossia andando oltre le indicazioni contenute nel foglietto illustrativo stesso.
Durante lo studio citato, condotto su 1208 pazienti affetti da AMD neovascolare, “sono state somministrate iniezioni intravitreali di ranibizumab o di bevacizumab in base a una cadenza mensile oppure al bisogno con valutazione mensile. Il risultato primario è stato un cambiamento medio nell’acuità visiva a un anno, con un limite non inferiore di 5 lettere [guadagnate] sull’ottotipo”. Come risultati – prosegue il New England Journal of Medicine – bevacizumab somministrato mensilmente è stato equivalente al ranibizumab somministrato mensilmente, rispettivamente con 8,0 e 8,5 lettere guadagnate“. Inoltre, proseguono i ricercatori, “la diminuzione media dello spessore retinico centrale è stata maggiore nel gruppo del ranibizumab mensile (196 µm) rispetto agli altri gruppi (da 152 a 168 µm)”. In conclusione, si legge ancora sulla prestigiosa pubblicazione britannica, “ad un anno il bevacizumab e il ranibizumab hanno avuto effetti equivalenti sull’acuità visiva quando somministrati secondo lo stesso protocollo. Il ranibizumab somministrato al bisogno, con una valutazione mensile, ha avuto effetti sulla visione equivalenti a quelli del ranibizumab somministrato mensilmente”. Tuttavia, conclude il CATT, “le differenze nell’incidenza di seri effetti collaterali richiedono ulteriori studi”.
Il 2 maggio 2012 la rivista Ophthalmology(rivista dell’Accademia Americana di Oftalmologia) ha pubblicato on-line uno studio (a firma degli stessi ricercatori del CATT) in cui si concludeva quanto segue: “In un periodo di due anni il ranibizumab e il bevacizumab hanno effetti simili sull’acuità visiva”. Lo studio – multicentrico (condotto in diverse università americane e presso il National Eye Institute statunitense) e randomizzato (ossia il trattamento è stato scelto casualmente) – è stato condotto su 1107 pazienti affetti da degenerazione maculare correlata all’età di tipo neovascolare. Anche l’American Academy of Ophthalmology ha dato risalto alla notizia nel proprio sito ufficiale, specificando tra l’altro che “nello studio biennale i tassi di gravi eventi come l’ictus, l’infarto e il decesso sono stati simili in chi ha ricevuto uno dei due farmaci”. Come avvenuto il primo anno – scrive ancora l’AAO – anche nel secondo si è manifestata una percentuale più alta di effetti collaterali non specifici gravi nei pazienti a cui veniva somministrato bevacizumab (40%) rispetto a quelli trattati con ranibizumab (32%). “I ricercatori, osserva ancora l’American Academy of Ophthalmology, sostengono che l’importanza degli effetti collaterali non è chiara; tuttavia potrebbe essere correlata al fatto che l’età media dei pazienti del CATT era di 80 anni, una fascia della popolazione in cui le malattie croniche o acute sono più comuni e ci si attende un tasso di ospedalizzazione più elevato”.
In un altro studio pubblicato nel 2013 sempre su Ophthalmologyi ricercatori scrivono che entrambi i principi attivi (ranibizumab e bevacizumab) “sono trattamenti molto efficaci nel preservare l’acuità visiva (VA) nelle persone con degenerazione maculare correlata all’età (AMD)” [6].
Nelle conclusioni di una ricerca pubblicata on-line su Retina il 7 agosto 2014 si legge: “Il bevacizumab e il ranibizumab hanno avuto un’efficacia equivalente sulla migliore acuità visiva corretta nel trattamento della degenerazione maculare legata all’età. Il ranibizumab ha avuto la tendenza a consentire un miglior risultato anatomico. Non ci sono state differenze tra i due farmaci nei tassi di mortalità, di eventi aterotrombotici o di eventi trombotici venosi”; ma saranno necessari ulteriori studi per averne ulteriori conferme [7].
Evoluzione dei farmaci anti-VEGF
EVOLUZIONE DEI FARMACI ANTI-VEGF
Il primo farmaco anti-VEGF ad essere introdotto e approvato dalla Food and Drug Administration (FDA) è stato il Pegaptanib nel 2004. Tuttavia, in relazione alla limitata percentuale di pazienti con un significativo miglioramento dell’acuità visiva, questo trattamento è stato poi superato dall’uso di farmaci più efficaci, quali Ranibizumab, Bevacizumab e Aflibercept. La molecola del Ranibizumab si è dimostrata essere piuttosto efficace in termini di mantenimento dell’acuità visiva del paziente, garantendo inoltre un’elevata sicurezza locale e sistemica. Il Bevacizumab è una molecola più grande, utilizzata comunque per il trattamento della AMD neovascolare come molecola alternativa e più economica rispetto ad altre. Il trattamento con i suddetti farmaci intravitreali prevede un’iniezione al mese per i primi 3 mesi, ed eventuali successive somministrazioni in base alla risposta del paziente. Una relativamente nuova risorsa è rappresentata dall’Aflibercept, molecola per la quale è suggerito un regime di trattamento con iniezioni intravitreali mensili per i primi tre mesi, seguite da iniezioni ogni 8 settimane. Sempre nell’ambito dei farmaci anti-VEGF, come nuova cura (ottobre 2019) per il trattamento della AMD neovascolare, è stato approvato da parte della FDA il Brolucizumab. Tale approvazione è poi giunta anche per l’Europa nel 2020. Si tratta di una nuova molecola, che si è dimostrata di pari efficacia rispetto all’Aflibercept, per il cui utilizzo è prevista sempre una somministrazione intravitreale mensile per i primi tre mesi (dose di carico), per poi passare ad un intervallo di trattamento ogni 12 settimane.
Infine, a Settembre 2022 la Commissione europea (CE) ha approvato Faricimab come nuovo farmaco per il trattamento della AMD umida. Il consenso informato della SOI cita: “Il trattamento della degenerazione maculare senile umida con Faricimab prevede iniezioni intravitreali ripetute a distanza di un mese per i primi 4 mesi. Successivamente, sulla base degli esiti anatomici e/o visivi a giudizio del medico oculista tra 20 e 24 settimane dopo l’inizio del trattamento, si raccomanda una valutazione dell’attività della malattia in modo da poter personalizzare la terapia. Nei pazienti senza attività di malattia, deve essere considerata la possibilità di somministrare Faricimab ogni 16 settimane (4 mesi). Nei pazienti con attività di malattia, deve essere considerato il trattamento ogni 8 settimane (2 mesi) o 12 settimane (3 mesi). Esistono dati di sicurezza limitati per intervalli tra le iniezioni pari o inferiori a 8 settimane”.
Prospettive future di trattamento dell’AMD
Una delle possibili prospettive future della terapia potrà essere basata su studi a carattere genetico. Inoltre, molto promettente è l’impiego di cellule staminali (in particolare quelle adulte riprogrammate), con cui negli anni a venire si potrà probabilmente rigenerare la retina. Risultati incoraggianti sono stati ottenuti, ad esempio, nel caso dell’AMD secca negli USA (approfondisci).
Altri gruppi di ricerca si stanno concentrando sugli accumuli proteici dannosi per la retina (drusen), cercando di rendere più efficiente la loro rimozione (autofagia). Tali meccanismi, tuttavia, necessitano di ulteriori studi. Bisogna però considerare che, essendo l’AMD una malattia retinica dovuta a diversi fattori, una cura risolutiva (specialmente per la forma secca) non è facile da mettere a punto.
Non esiste dunque ad oggi una cura definitiva per la maculopatia secca, tuttavia nuove tecnologie stanno emergendo, per rallentare la progressione della malattia. Una di queste è la iontoforesisclerale, una procedura innovativa che consente di rilasciare dentro l’occhio, tramite un piccolo dispositivo medico elettrico, la luteina. Quest’ultima è una sostanza naturale appartenente al gruppo dei carotenoidi in grado di formare una sorta di schermo protettivo sulle cellule retiniche, favorendone l’integrità e la corretta funzionalità. La luteina non può essere prodotta dal nostro organismo, pertanto l’unica fonte di assunzione sono i cibi o gli integratori alimentari specifici (ampiamente utilizzati per molti anni come unico trattamento per la maculopatia secca, come già accennato in precedenza). Il problema dell’assunzione orale della luteina consiste nel fatto che, una volta ingerita per via orale, viene eliminata per la maggior parte e soltanto una quantità minima riesce effettivamente ad accumularsi nella macula. Lo scopo del trattamento di iontoforesi è quello di riuscire a somministrare per via topica la luteina e veicolarne direttamente nella macula una concentrazione maggiore. Il trattamento è piuttosto semplice, veloce, indolore e viene eseguito in ambulatorio, si effettua attraverso l’applicazione di un elettrodo sulla fronte ed un conetto di plastica posizionato sull’occhio (dopo aver instillato delle gocce di collirio anestetico), contenente la soluzione di luteina. La durata del trattamento è di pochi minuti, può essere ripetuto, non presenta particolari effetti collaterali (per lo più i pazienti possono sviluppare un lieve arrossamento oculare e/o secchezza transitoria). Rara la possibilità di avere problemi di disepitelizzazione della cornea. Riassumendo, la iontoforesi sclerale rappresenta un approccio nuovo per il trattamento della degenerazione maculare secca, riuscendo a garantire una somministrazione più mirata della luteina, con un’efficacia potenzialmente superiore rispetto all’integrazione orale; saranno necessari studi più ampi per confermare i risultati e l’efficacia a lungo termine di tale metodica.
In ambito riabilitativo, si sta utilizzando con un certo successo la tecnica del biofeedback, che consiste nella fotostimolazione neuronale dei recettori retinici attraverso degli spot di luce ad una particolare frequenza, emessi da dispositivi dedicati. Il biofeedback, viene effettuato nella riabilitazione dei pazienti con maculopatia in modo da aumentare la stabilità della fissazione e potenziare l’acutezza visiva. Utili anche gli ausili per gli ipovedenti quali i videoingranditori (disponibili oggi in molti centri ospedalieri). E’ importante diagnosticare precocemente i primi sintomi della degenerazione maculare legata all’età, in modo da attuare le misure preventive più idonee. Tuttavia i pazienti devono comprendere che si tratta di una patologia degenerativa e che, come tale, può lentamente aggravarsi nel tempo; talvolta tutte le terapie effettuate potrebbero non essere sufficienti e, quindi, è il caso di non avere aspettative eccessive.
UN CORRETTO STILE DI VITA: ALIMENTAZIONE VARIA, NIENTE FUMO E PIU’ ESERCIZIO FISICO
Con l’aumento dell’incidenza della degenerazione maculare legata all’età è cresciuto anche il numero degli studi effettuati.
Eliminare il fumo è la prima buona pratica di vita. Inoltre controlli accurati del sistema cardiovascolare sono assolutamente raccomandabili. È stato stimato che in chi fuma il rischio di essere colpiti da AMD aumenta fino a tre volte. [14]
Non va assolutamente trascurato l’esercizio fisico moderato, importante ad ogni età. Numerosi studi hanno ipotizzato, infatti, che per chi lo pratica regolarmente sia più difficile essere colpiti dalla degenerazione maculare legata all’età o, se questo avviene, la sua evoluzione è generalmente più lenta. Infine, non bisogna assolutamente trascurare il fatto che i raggi ultravioletti possano contribuire a danneggiare la macula: specialmente se si sono avuti altri casi di AMD in famiglia, bisogna prestare più attenzione al corretto uso di occhiali scuri con lenti a norma di legge (vedi consigli utili). Uno dei principali fattori di rischio potrebbe proprio essere l’esposizione prolungata e cumulativa ai raggi ultravioletti, oltre naturalmente all’età, dunque può essere importante far uso di filtri di buona qualità.
[1] Nei familiari di primo grado malati di AMD il rischio di svilupparla è superiore, rispetto alla popolazione generale, dalle 3 alle 6 volte. Esistono indicativamente cinque categorie di geni coinvolti: quelli che controllano l’infiammazione e la risposta cellulare, il metabolismo e il trasporto dei lipidi, la matrice extracellulare e l’adesione cellulare, l’angiogenesi e la risposta allo stress cellulare.
[3] L’Avastin, originariamente sintetizzato come principio attivo di un trattamento antitumorale (contro il cancro del colon retto), è stato poi impiegato anche per altri scopi, compresi quelli oftalmici, vista la sua proprietà di inibire la proliferazione incontrollata dei vasi retinici dannosi. Viene utilizzato esclusivamente a scopi di ricerca; ad uso oftalmico è impiegato in Italia solo come off-label, ossia esulando delle indicazioni terapeutiche riportate nel foglietto illustrativo.
[5] “Ranibizumab and Bevacizumab for Neovascular Age-Related Macular Degeneration”, The CATT Research Group, N Engl J Med. 2011 Apr 28., e-pub. Lo studio è stato finanziato dal National Eye Institute (ClinicalTrials.gov number, NCT00593450).
[6] Glenn J. Jaffe, Daniel F. Martin, Cynthia A. Toth, Ebenezer Daniel, Maureen G. Maguire, Gui-Shuang Ying, Juan E. Grunwald, Jiayan Huang, “Macular Morphology and Visual Acuity in the Comparison of Age-related Macular Degeneration Treatments Trials”, Comparison of Age-related Macular Degeneration Treatments Trials Research Group, Ophthalmology– September 2013 (Vol. 120, Issue 9, Pages 1860-1870, DOI: 10.1016/j.ophtha.2013.01.073)
[7] Chen G, Li W, Tzekov R, Jiang F, Mao S, Tong Y, “Bevacizumab versus ranibizumab for neovascular age-related macular degeneration: a meta-analysis of randomized controlled trials”, Retina, 2014 Aug 7 (Epub ahead of print)
[8] Si veda ad esempio: Evans JR et al, “28,000 cases of age related macular degeneration causing visual loss in people aged 75 years and above in the UK may be attributable to smoking”, British Journal of Ophthalmology (2005)
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Pagina pubblicata il 31 luglio 2007. Ultima revisione scientifica: 11/09/2025
Contatta l’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Il Numero Verde di consultazione oculistica è attivo dal lunedì al venerdì dalle 10 alle 13. Scrivi nel Forum: un medico oculista ti risponderà gratuitamente.
La retinopatia miopica è una patologia della retina tipica delle persone affette da miopia elevata (errore refrattivo > – 6 diottrie o lunghezza assiale > 26,5 mm). La gravità delle alterazioni retiniche aumenta al crescere del vizio refrattivo. Alcune alterazioni tipiche di questi occhi si possono trovare, comunque, anche in persone con una lieve miopia o persino emmetropi (prive di difetti visivi); dunque sono sempre consigliabili visite oculistiche periodiche di controllo.
Da cosa è causata?
Nella miopia l’occhio è più allungato rispetto alla norma, proporzionalmente al difetto refrattivo, provocando una serie di alterazioni anatomiche, a carico di sclera, coroide, retina e disco ottico.. La retina, in particolare, subisce degli stiramenti con formazione di lesioni in periferia (piccoli fori, anche microscopici), in quanto non riesce ad adattarsi bene all’allungamento del bulbo. Tale processo di “stiramento” coinvolge anche la coroide, provocando un assottigliamento del complesso vascolare coroideale e rendendo il tessuto retinico meno irrorato per mezzo dei vasi sanguigni. L’ipossia retinica, in seguito, può portare al rilascio del fattore di crescita endoteliale vascolare, che fa da mediatore per lo sviluppo di neovasi a livello della coroide (CNV). A livello della retina periferica, il processo di assottigliamento del bulbo, può indurre tutta una serie di processi degenerativi, con formazione di piccoli fori, rotture, fino ad arrivare alla complicanza più grave: il distacco di retina.
A che età può presentarsi?
In genere la miopia insorge in età scolare, aumenta nel periodo dello sviluppo e tende a stabilizzarsi intorno ai 20-25 anni, aumentando solo lievemente dopo quell’età. La miopia patologica o degenerativa, invece, insorge già nei bambini piccoli (tipicamente a 2-3 anni d’età) e progredisce col passare degli anni arrivando anche a valori molto elevati (ad esempio 30 diottrie), poiché il bulbo oculare continua ad allungarsi in modo patologico (anche al termine del periodo di crescita), arrivando a compromettere l’integrità di tutte le strutture anatomiche. Purtroppo, nella maggior parte dei casi, tutta la serie di processi descritti relativi alla miopia patologica, conducono ad un importante deterioramento dell’ acuità visiva, colpendo spesso i pazienti durante l’età lavorativa e impedendo loro di svolgere anche le più semplici attività quotidiane.
Ci sono forme di prevenzione?
La miopia è dovuta sia a fattori ereditari che a fattori ambientali. È necessario, quindi, far crescere i bambini all’aria aperta, evitare che leggano o giochino in condizioni di scarsa luminosità e bisogna seguire uno stile alimentare (a tutte le età), prediligendo le sostanze antiossidanti. L’assunzione d’integratori alimentari retinici – tra cui la mirtillina – rappresenta una forma di prevenzione importante soprattutto per la protezione dei vasi.
Quali sono le terapie esistenti?
Non esiste una terapia medica che curi la retinopatia miopica. Al contrario, si possono curare le complicanze che insorgono nella retina di un occhio miope, intervenendo in base alle degenerazioni o lesioni che si vengono ad instaurare. Le rotture, i fori e le aree di maggior assottigliamento retinico (degenerazioni regmatogene) vengono trattate con il laser argon. Contro la formazione di nuovi vasi sottoretinici in zona maculare (CNV), la terapia fotodinamica (PDT) si è dimostrata efficace, soprattutto in passato, così come anche (più di recente) l’utilizzo di iniezioni intravitreali di farmaci detti antiangiogenici (o anti-VEGF, che inibiscono la proliferazione di vasi retinici indesiderati).
Si può diventare ciechi a causa della retinopatia miopica?
No, anche se può comportare una minorazione visiva rilevante a causa delle sue complicanze (come la neovascolarizzazione coroideale, detta CNV). La CNV può avere un’evoluzione lenta; infatti, dopo un brusco peggioramento iniziale della vista si può assistere a un miglioramento dovuto alla riduzione dei fenomeni emorragici ed essudativi e all’instaurarsi di una fissazione non più centrale, mantenendo quindi una discreta visione anche al termine della sua evoluzione.
Quando il processo di neovascolarizzazione va incontro a cicatrizzazione si può osservare uno scotoma (macchia scura) che col tempo può allargarsi, con conseguente diminuzione della vista sino alla cecità legale ). Altra causa di minorazione visiva può essere lo stafiloma miopico, generalmente presente in giovane età, la cui gravità può aumentare negli anni; la prognosi visiva dipende però dalla sua localizzazione. Infatti, quando lo stafiloma è centrale, nel 50% dei casi il paziente diventa legalmente cieco dopo i 60 anni.
Alterazioni dovute alla retinopatia miopica:
Stafiloma miopico: si osserva con l’esame del fondo oculare. È localizzato a livello della zona posteriore del bulbo (polo posteriore). È presente in giovane età e consiste in uno sfiancamento posteriore del bulbo, che lascia intravedere i vasi sanguigni della sottostante coroide (strato intermedio dell’occhio posto tra la retina e la sclera); si può associare a un’atrofia dell’epitelio pigmentato retinico (strato esterno della retina neurosensoriale, attaccato alla coroide, che nutre le cellule visive).
Distrofia e atrofia corioretinica: è causata dall’assottigliamento dell’epitelio pigmentato retinico che rende visibile la vascolarizzazione coroideale. Si creano, in pratica, delle isole di atrofia corio-capillare completa, più o meno confluenti e attraversate da vasi coroideali profondi ben visibili sullo strato bianco-giallastro della sclera. In base alla loro localizzazione (in particolare se coinvolgono la fovea), sono causa di deterioramento funzionale più o meno grave, sino a poter indurre anche l’abolizione completa della visione centrale.
Tilted disc: è caratterizzato da uno stafiloma decentrato. Il nervo ottico appare di forma ovale con un maggiore asse orizzontale. Si può riscontrare anche in miopie inferiori a 6 diottrie, ma spesso si associa a un astigmatismo.
Rotture membrana di Bruch: si evidenziano maggiormente negli occhi con una lunghezza assiale superiore a 26 mm. All’esame del fondo oculare appaiono come linee giallastre. Possono essere prive di sintomi o si possono accompagnare ad emorragie sottoretiniche tondeggianti, piccole e a contorni ben definiti. Se presenti a livello foveale (zona centrale della macula) provocano una riduzione della vista.
Neovascolarizzazione coroideale (CNV): è la complicanza più temibile. Si presenta come una lesione grigiastra al polo posteriore come conseguenza di una rottura della membrana di Bruch. È caratterizzata da essudazione ed emorragie retinica. Provoca visione distorta, annebbiamento e scotoma centrale (zona di non visione al centro del campo visivo).
Distacco di retina: può essere posteriore, partendo da un foro maculare o si può originare da una rottura retinica periferica.
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus Leggi le condizioni generali di consultazione di questo sito
Pagina pubblicata il 14 maggio 2009. Ultimo aggiornamento:15/12/2022
Ultima revisione scientifica: 15/12/2022
Contatta l’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Il Numero Verde di consultazione oculistica è attivo dal lunedì al venerdì dalle 10 alle 13. Scrivi nel Forum: un medico oculista ti risponderà gratuitamente.
Il retinoblastoma è il tumore maligno oculare più frequente in età pediatrica e rappresenta il 4% dei tumori pediatrici. Statisticamente colpisce annualmente la retina di un bambino ogni ventimila nati vivi; può svilupparsi sia in un solo occhio (60% dei casi) che in entrambi (40% dei casi). I primi segni e sintomi sono riscontrati in bambini di età inferiore ai 3 anni; le diagnosi dopo i 6 anni sono piuttosto rare. Nel 40% dei casi ha un’origine ereditaria. In Italia, come nel resto dei paesi industrializzati, la sopravvivenza dei pazienti affetti da retinoblastoma supera il 95%, mentre nei paesi in via di sviluppo si aggira intorno al 50%. Tale differenza è strettamente correlata ad una diagnosi precoce e all’utilizzo di trattamenti adeguati, maggiormente disponibili nei paesi più industrializzati.
Da cosa è causato?
Da una mutazione del gene oncosoppressore RB1, che comporta la proliferazione incontrollata di cellule e lo sviluppo del tumore all’interno dell’occhio. La mutazione può essere trasmessa per via ereditaria o insorgere spontaneamente (mutazione sporadica).
Quali sono i sintomi?
Purtroppo, è in genere privo di sintomi, per cui spesso viene diagnosticato tardivamente. Il segno più frequente è la leucocoria ovvero un riflesso bianco nella pupilla simile a una piccola macchia dovuto alla massa tumorale che si sviluppa all’interno della camera vitrea. Frequente è anche lo strabismo (la deviazione di uno o entrambi gli occhi verso l’interno o verso l’esterno).
Come si effettua la diagnosi?
Inizialmente attraverso l’esame del fondo oculare. In seconda battuta è fondamentale che i bambini siano sottoposti ad ecografia oculare; talvolta, nei centri ad alta specializzazione vengono sottoposti anche a fluorangiografia. È importante sempre che gli esami siano eseguiti in tutti e due gli occhi, in quanto le forme inizialmente unilaterali possono poi coinvolgere anche l’altro occhio. Il bambino deve essere sottoposto, se possibile, a TAC e Risonanza Magnetica per valutare il coinvolgimento del nervo ottico e/o la presenza di eventuali metastasi.
Quali sono le terapie disponibili?
Attualmente esistono dei protocolli terapeutici stabiliti dalla comunità scientifica che impongono determinati trattamenti a seconda dello stadio della malattia. I trattamenti locali includono: fotocoagulazione laser, crioterapia, termoterapia transpupillare e brachiterapia (applicazione di placche radioattive). A questi trattamenti viene associata attualmente una chemioterapia per via sistemica. Negli ultimi tempi si sta affermando l’utilizzo di chemioterapia per via arteriosa, attraverso l’arteria oftalmica. Purtroppo nelle forme più avanzate di malattia è necessario rimuovere chirurgicamente il bulbo oculare malato (enucleazione).
Con che frequenza bisogna fare visitare i malati?
I bambini affetti da retinoblastoma devono essere seguiti con controlli in tempi molto ravvicinati per via della velocità di progressione della malattia e, anche dopo essere stati curati, devono essere sottoposti a controlli semestrali per i primi 5 anni dalla fine della terapia.
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus Leggi le condizioni generali di consultazione di questo sito
Pagina pubblicata il 12 dicembre 2007. Ultimo aggiornamento: 15 febbraio 2019.
Ultima revisione scientifica:02/11/2022
Contatta l’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Il Numero Verde di consultazione oculistica è attivo dal lunedì al venerdì dalle 10 alle 13. Scrivi nel Forum: un medico oculista ti risponderà gratuitamente.
L’esame del fondo oculare (detto anche fundus oculi o oftalmoscopia) è un esame diagnostico che viene utilizzato per visualizzare le strutture interne del bulbo, ovvero il corpo vitreo, la testa del nervo ottico, la retina centrale (la macula) e la retina periferica.
Come si esegue?
Si effettua dopo aver dilatato la pupilla mediante instillazione di speciali colliri (detti midriatici). Si esegue in ambiente scarsamente illuminato, seduti o distesi. Si tratta di un esame non invasivo né doloroso, il paziente potrebbe però avvertire un lieve fastidio, dovuto all’abbagliamento provocato dal fascio di luce indirizzato nell’occhio durante l’esplorazione.
Con quali strumenti si effettua?
Con uno strumento chiamato “oftalmoscopio”.
L’oftalmoscopio diretto è un apparecchio simile ad una torcia, munito di una sorgente luminosa posizionata nella testina e di un sistema di lenti in grado di correggere eventuali vizi refrattivi del paziente o dell’esaminatore. Consente di ottenere un’immagine del fondo oculare diritta (non capovolta), con un discreto ingrandimento e buona percezione dei dettagli; il campo d’osservazione non è molto ampio, consente infatti una buona visualizzazione della parte centrale della retina, non si riesce invece ad apprezzare la periferia retinica. L’oftalmoscopio indiretto è uno strumento che viene posizionato sul capo dell’esaminatore, provvisto a sua volta di un sistema di illuminazione. Consente di avere una visualizzazione della retina molto più ampia, così da osservare bene anche la periferia, offre però un’immagine meno ingrandita e invertita. Inoltre, con l’oftalmoscopio indiretto si utilizza una lente tra l’apparecchio e l’esaminato. Anche con la lampada a fessura è possibile eseguire l’esame del fondo oculare utilizzando lenti addizionali, posizionate tra lo strumento e l’occhio del paziente.
Quando serve?
Tutte le persone si devono sottoporre all’esame del fondo dell’occhio con una periodicità che dipende dall’età, dalla presenza di eventuali patologie sistemiche e/o oculari.
Persone che non hanno alcun disturbo e hanno meno di 40 anni possono sottoporsi all’esame una volta ogni 18-24 mesi. Potrebbe bastare una volta l’anno per chi è affetto da miopia o da patologie oculari non gravi (oppure per individui sani con più di 40 anni).
Chi ha problemi retinici o di diabete e ipertensione dovrà sottoporsi a controlli ravvicinati, la cui cadenza esatta deve essere decisa dal singolo oculista a seconda della situazione specifica.
Comunque è importante sottoporsi a un esame del fondo oculare anche quando si vedono per la prima volta dei fosfeni (lampi luminosi) o miodesopsie (mosche volanti). Nel primo caso il corpo vitreo potrebbe esercitare una trazione sulla retina, con rischio di un suo distacco.
Cosa consente di osservare?
L’esame del fondo oculare permette di osservare lo stato del corpo vitreo (il gel che riempie il bulbo oculare) e sue eventuali degenerazioni e di visualizzare l’albero vascolare arterioso e venoso retinico. Permette, inoltre, di osservare la macula, la zona centrale della retina che consente la visione distinta, in grado di farci percepire chiaramente i dettagli degli oggetti, di leggere i caratteri piccoli, riconoscere i volti delle persone. Eventuali degenerazioni e anomalie (distrofie) della macula possono essere dunque diagnosticate e monitorate con l’esame del fondo oculare. Si può, infine, valutare la conformazione della testa del nervo ottico individuando eventuali patologie (locali o sistemiche).
Quali malattie?
Il diabete o l’ipertensione sono patologie che colpiscono i vasi: ciò che accade nell’occhio avviene, ad esempio, anche nel rene e nel cuore. Il vantaggio è che, con il fondo dell’occhio, si riescono a visualizzare le vene e le arterie con sistemi non invasivi. Per quanto riguarda le patologie oculari il semplice esame del fondo dell’occhio ci permette di diagnosticare e monitorare nel tempo diverse patologie quali: maculopatie, retinite pigmentosa, distacco di retina, neuriti ottiche, occlusioni venose e arteriose retiniche, distacco posteriore del vitreo, emorragie vitreali, glaucoma. In questo modo l’oculista può impostare, in maniera tempestiva, una terapia mirata, per scongiurare che si sviluppino danni gravi e irreversibili per i nostri occhi.
Quali sono le controindicazioni di un esame del fondo oculare?
Generalmente non ha controindicazioni. Tuttavia, bisogna stare attenti se si ha la camera anteriore dell’occhio (spazio compreso tra l’iride e la cornea) poco profonda. Instillando le gocce midriatiche (per dilatare la pupilla), si corre il rischio di provocare la chiusura dell’angolo irido-corneale e di ostacolare così il normale deflusso dell’umore acqueo, provocando un innalzamento improvviso ed importante della pressione oculare. Ecco perché, in tali casi, l’esame del fondo oculare potrebbe essere effettuato con la pupilla non dilatata, riuscendo ad esplorare in tal modo esclusivamente il polo posteriore della retina.
Inoltre possono presentarsi reazioni avverse ai colliri che si usano per la dilatazione: possono andare da una semplice reazione allergica a fenomeni sistemici più gravi. Tali effetti sono molto rari e, comunque, controllabili. I midriatici (gocce per dilatare le pupille) sono molti; nel caso in cui vi siano effetti collaterali è bene ricordare quale tipo di farmaco abbia creato problemi, comunicandolo tempestivamente all’oculista o recandosi a un pronto soccorso (possibilmente oculistico). Infine bisogna far passare un lasso di tempo sufficiente per riacquistare la visione che precedeva la dilatazione della pupilla: non ci si può mettere subito alla guida per motivi di sicurezza, dato che si vede annebbiato (leggi anche i consigli utili).
Si può fotografare il fondo dell’occhio?
Sì, con uno strumento chiamato “retinografo”. La retinografia è appunto un esame che permette di documentare lo stato del fondo oculare grazie a immagini digitali a colori, senza dover dilatare la pupilla.








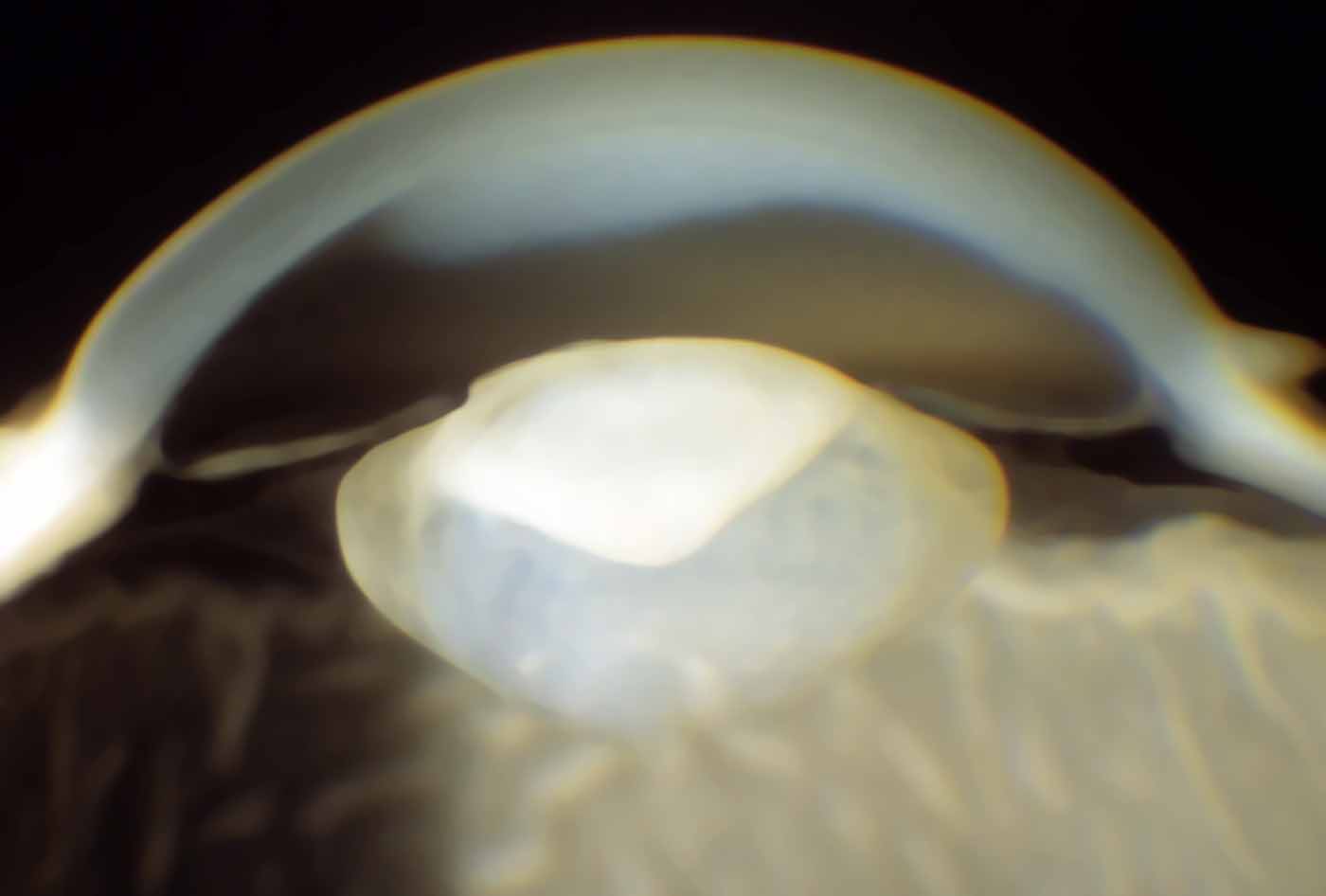 Si tratta di una patologia che colpisce il
Si tratta di una patologia che colpisce il  Sì, generalmente è trattabile con un’operazione chirurgica. Ovviamente alcune cataratte infantili che non interferiscono sulla capacità visiva, in quanto piccole o poco dense, non richiedono la chirurgia.
Sì, generalmente è trattabile con un’operazione chirurgica. Ovviamente alcune cataratte infantili che non interferiscono sulla capacità visiva, in quanto piccole o poco dense, non richiedono la chirurgia.






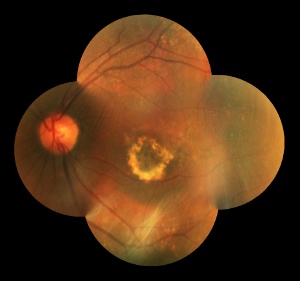 capillare, tipiche dell’età avanzata: la forma secca (o atrofica) e quella umida (o essudativa); queste andrebbero considerate come due patologie distinte, poiché le loro prognosi ed eventuali terapie sono del tutto diverse.
capillare, tipiche dell’età avanzata: la forma secca (o atrofica) e quella umida (o essudativa); queste andrebbero considerate come due patologie distinte, poiché le loro prognosi ed eventuali terapie sono del tutto diverse.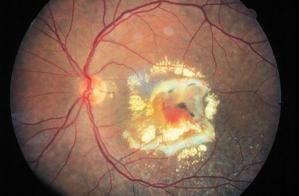
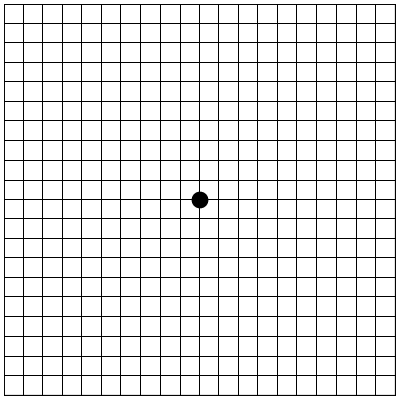
 La retinopatia miopica è una patologia della
La retinopatia miopica è una patologia della 



 Il diabete o l’ipertensione sono patologie che colpiscono i vasi: ciò che accade nell’occhio avviene, ad esempio, anche nel rene e nel cuore. Il vantaggio è che, con il fondo dell’occhio, si riescono a visualizzare le vene e le arterie con sistemi non invasivi. Per quanto riguarda le patologie oculari il semplice esame del fondo dell’occhio ci permette di diagnosticare e monitorare nel tempo diverse patologie quali: maculopatie, retinite pigmentosa, distacco di retina, neuriti ottiche, occlusioni venose e arteriose retiniche, distacco posteriore del vitreo, emorragie vitreali, glaucoma. In questo modo l’oculista può impostare, in maniera tempestiva, una terapia mirata, per scongiurare che si sviluppino danni gravi e irreversibili per i nostri occhi.
Il diabete o l’ipertensione sono patologie che colpiscono i vasi: ciò che accade nell’occhio avviene, ad esempio, anche nel rene e nel cuore. Il vantaggio è che, con il fondo dell’occhio, si riescono a visualizzare le vene e le arterie con sistemi non invasivi. Per quanto riguarda le patologie oculari il semplice esame del fondo dell’occhio ci permette di diagnosticare e monitorare nel tempo diverse patologie quali: maculopatie, retinite pigmentosa, distacco di retina, neuriti ottiche, occlusioni venose e arteriose retiniche, distacco posteriore del vitreo, emorragie vitreali, glaucoma. In questo modo l’oculista può impostare, in maniera tempestiva, una terapia mirata, per scongiurare che si sviluppino danni gravi e irreversibili per i nostri occhi.