

Retinite pigmentosa
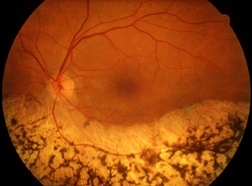
La retinite pigmentosa è una malattia genetica rara che colpisce la retina di entrambi gli occhi, causando un progressivo deterioramento della funzione visiva. Fa parte di un insieme di patologie ereditarie caratterizzate dalla degenerazione graduale delle cellule retiniche responsabili della percezione della luce. I primi sintomi riguardano generalmente la difficoltà a vedere in condizioni di scarsa illuminazione e la riduzione della visione periferica. Con il passare del tempo, la malattia può interessare anche la visione centrale, compromettendo in modo significativo la capacità visiva complessiva. Conosciuta anche come retinosi pigmentaria o retinopatia pigmentosa, questa patologia determina un progressivo restringimento del campo visivo e una diminuzione dell’acuità visiva. Negli stadi più avanzati può portare a ipovisione severa e, nei casi più gravi, alla cecità.
Come funziona la retina?
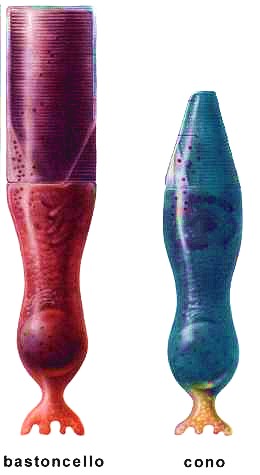
La retina è un sottile strato di tessuto nervoso organizzato in più livelli, rappresenta la componente fondamentale dell’occhio, perché svolge un ruolo centrale nel meccanismo della visione. La luce proveniente dall’ambiente esterno viene focalizzata su questa struttura, dove viene convertita in impulsi elettrochimici. Tali segnali vengono poi trasmessi al cervello attraverso il nervo ottico e qui elaborati fino ad essere convertiti in immagini. Quando le cellule retiniche subiscono un danno, questa catena di trasmissione si interrompe e la capacità visiva risulta compromessa. All’interno della retina sono presenti circa 131 milioni di cellule fotosensibili, chiamate fotorecettori, che si suddividono in due categorie principali:
- I coni, così denominati per la loro forma, sono responsabili della percezione dei dettagli e dei colori. Ne esistono tre varietà, ciascuna sensibile a una specifica lunghezza d’onda corrispondente al rosso, al verde e al blu; la combinazione delle loro risposte viene interpretata dal cervello come una determinata tonalità cromatica. I coni sono circa 6 milioni e si concentrano soprattutto nella zona centrale della retina, chiamata macula. Per questo motivo risultano indispensabili per la visione centrale, necessaria ad attività come la lettura, il riconoscimento dei volti e la guida. La loro capacità di discriminare i particolari è enormemente superiore a quella dei bastoncelli.
- I bastoncelli, invece, hanno una forma più allungata e rispondono principalmente alle variazioni di luminosità e al movimento. Sono circa 125 milioni e si distribuiscono prevalentemente nelle aree periferiche della retina, dove superano di gran lunga il numero dei coni. Nella regione centrale sono poco presenti, mentre diventano molto numerosi verso la periferia. Grazie ai bastoncelli siamo in grado di percepire oggetti in movimento ai margini del campo visivo, anche senza distinguerne con precisione i dettagli.


Quante persone colpisce la retinite pigmentosa?
La retinite pigmentosa è una malattia rara che colpisce circa una persona ogni 4.000 negli Stati Uniti ed è ancora oggi una delle principali cause di riduzione grave della vista e di cecità nelle persone sotto i 60 anni. In Italia, secondo i dati disponibili, si stima che ne siano affette circa 30.000 persone, mentre in Europa il numero sale a circa 167.000. Se si considerano nel loro insieme tutte le degenerazioni ereditarie della retina, il totale dei soggetti coinvolti a livello mondiale raggiungerebbe i 2 milioni di cui circa 1,5 milioni soffrirebbero proprio di retinite pigmentosa.Attualmente non esiste ancora una cura in grado di risolvere definitivamente la malattia. Tuttavia, numerosi gruppi di ricerca stanno lavorando su possibili terapie, con l’obiettivo di rallentarne l’evoluzione o migliorare la qualità della vita delle persone colpite. La retinite pigmentosa può manifestarsi in età molto diverse: nella maggior parte dei casi compare tra l’adolescenza e l’età adulta, ma non mancano situazioni in cui i primi sintomi si presentano già durante l’infanzia. L’andamento della malattia è generalmente progressivo: la capacità visiva tende a diminuire nel tempo, con un peggioramento graduale che può diventare significativo negli anni. L’unico aspetto sul quale la comunità scientifica concorda pienamente riguarda l’origine genetica della retinite pigmentosa. Si tratta infatti di una patologia ereditaria, trasmessa all’interno delle famiglie secondo modalità ormai ben note. Le ricerche condotte finora hanno permesso di individuare circa un centinaio di geni che, se alterati, possono essere responsabili dell’insorgenza della malattia, spiegando così la grande varietà di forme e gravità con cui essa si presenta.
Quali forme esistono di retinite pigmentosa?
La retinite pigmentosa è una malattia di origine genetica che può essere trasmessa secondo diverse modalità ereditarie. Le forme di trasmissione più comuni sono quella autosomica dominante, autosomica recessiva e quella legata al cromosoma X; in una parte dei casi la patologia si manifesta invece in modo apparentemente sporadico, senza altri casi noti all’interno della stessa famiglia.
- Nella trasmissione autosomica dominante, è sufficiente che uno dei due genitori sia affetto dalla malattia perché esista una probabilità del 50% che ciascun figlio erediti la mutazione responsabile. Questa forma colpisce maschi e femmine con la stessa frequenza.
- La forma autosomica recessiva si verifica quando entrambi i genitori sono portatori sani della mutazione genetica, pur non presentando sintomi. In questo caso, ogni figlio ha una probabilità del 25% di sviluppare la malattia. Anche questa modalità interessa in egual misura entrambi i sessi.
- Esiste poi la trasmissione legata al cromosoma X, nella quale il gene responsabile è localizzato sul cromosoma sessuale X. In questa situazione, la madre è generalmente portatrice sana e può trasmettere la mutazione ai figli maschi, che risultano affetti, con una probabilità del 50%. Le figlie femmine, invece, nella maggior parte dei casi non sviluppano la malattia, ma hanno a loro volta il 50% di probabilità di essere portatrici sane. Di conseguenza, questa forma di retinite pigmentosa colpisce quasi esclusivamente individui di sesso maschile.
- In circa il 30% dei casi la retinite pigmentosa viene definita sporadica, poiché si osserva un solo soggetto affetto all’interno della famiglia, anche considerando più generazioni. Tuttavia, questa definizione si basa esclusivamente sull’anamnesi familiare: non sempre è possibile escludere con certezza una trasmissione autosomica recessiva o legata al sesso, soprattutto quando la persona colpita è un maschio.
Infine, quando la retinite pigmentosa si presenta in associazione a una perdita dell’udito, si parla di sindrome di Usher, una condizione genetica complessa che coinvolge sia il sistema visivo sia quello uditivo.
Quali sono i sintomi?

I primi segnali che possono far sospettare la presenza di una retinite pigmentosa sono principalmente legati a difficoltà visive che compaiono in modo graduale. Uno dei sintomi più caratteristici è la ridotta capacità di vedere in condizioni di scarsa illuminazione, soprattutto di sera o di notte. Le persone colpite possono avere problemi a muoversi in ambienti poco illuminati o a guidare dopo il tramonto, e spesso riferiscono un adattamento lento quando si passa da spazi luminosi a luoghi più bui. Questo disturbo è dovuto al fatto che, nelle fasi iniziali della malattia, vengono colpiti soprattutto i bastoncelli, le cellule della retina responsabili della visione notturna. Con il progredire della patologia, si manifesta anche una progressiva riduzione del campo visivo. Diventa sempre più difficile percepire ciò che si trova ai lati, tanto che oggetti laterali, gradini o ostacoli bassi possono non essere notati. Questa condizione, spesso definita “visione tubulare”, tende ad accentuarsi nel tempo e, nei casi più avanzati, può estendersi fino a coinvolgere la parte centrale della retina, compromettendo anche la visione centrale. La velocità con cui si verifica questo peggioramento e l’età di comparsa dei sintomi variano da persona a persona e dipendono da diversi fattori, tra cui il tipo di alterazione genetica alla base della malattia. A questi disturbi si associano frequentemente una maggiore sensibilità alla luce, una riduzione della sensibilità al contrasto e una crescente difficoltà nel riconoscere con chiarezza l’ambiente circostante.
Come si effettua la diagnosi?
La diagnosi di retinite pigmentosa, quando sono presenti i sintomi tipici della malattia, è generalmente agevole ed è di competenza dell’oculista, spesso in collaborazione con il genetista. Il percorso diagnostico si basa su una serie di esami clinici e strumentali mirati a valutare la funzionalità e la struttura della retina, in particolare:
- l’esame del visus;
- l’osservazione del fondo oculare;
- lo studio del campo visivo;
- l’elettroretinogramma;
- la fluorangiografia retinica.
La malattia può essere identificata in diverse fasi della vita: dall’infanzia all’adolescenza, fino all’età adulta, anche se l’età di esordio varia notevolmente da persona a persona. Nei casi in cui il quadro clinico non sia immediatamente chiaro, la diagnosi si fonda su una valutazione approfondita di tutti gli elementi disponibili, come l’età di comparsa dei sintomi, la modalità di progressione della malattia e l’eventuale presenza di altri disturbi oculari o sistemici. In queste situazioni possono risultare particolarmente utili esami più specifici, come lo studio elettrofisiologico, che comprende l’elettroretinogramma (ERG) e l’elettrooculogramma (EOG), oltre ai test di adattamento al buio. Anche la valutazione della percezione dei colori e la fluorangiografia della retina possono fornire informazioni complementari. Un aspetto importante del processo diagnostico è inoltre l’analisi dell’intero nucleo familiare, necessaria per individuare il tipo di trasmissione ereditaria della malattia e definire correttamente il quadro genetico.
- L’esame del visus, consente di valutare l’acuità visiva a livello della parte centrale della retina. Viene eseguito chiedendo al paziente di leggere, su un apposito tabellone posizionato di solito ad una distanza di 3 o 5 metri, caratteri di dimensioni progressivamente più piccole.
- L’esame del fondo oculare ha lo scopo di analizzare la struttura della retina e di individuare l’eventuale presenza di depositi pigmentari sulla sua superficie. Nella retinite pigmentosa, tali alterazioni assumono spesso un aspetto caratteristico, comunemente descritto come “a spicole ossee”. È importante sottolineare, tuttavia, che alcune forme di retinite possono manifestare sintomi del tutto simili pur non presentando queste tipiche modificazioni del fondo oculare.
- L’esame del campo visivo permette di valutare la sensibilità della retina agli stimoli luminosi nelle diverse aree della retina. Fornisce una documentazione oggettiva delle difficoltà riferite dal paziente, in particolare durante il movimento, e rappresenta uno strumento essenziale per seguire nel tempo l’evoluzione della malattia.
- L’elettroretinogramma (ERG) è un esame che consente di registrare l’attività elettrica della retina in risposta a specifici stimoli luminosi. Attraverso questo test è possibile valutare in modo separato la funzionalità dei due principali tipi di fotorecettori, i coni e i bastoncelli. L’ERG riveste un ruolo fondamentale nella diagnosi della retinite pigmentosa, poiché già nelle fasi iniziali della malattia il tracciato risulta spesso marcatamente ridotto o quasi assente, indicando una compromissione precoce della funzione retinica.
- La fluorangiografia (FAG) è un esame diagnostico che prevede l’iniezione per via endovenosa di un colorante (fluoresceina), seguita dall’esecuzione di una serie di fotografie della retina in momenti successivi. Attraverso la circolazione sanguigna, il colorante raggiunge i vasi retinici, rendendo visibili arterie, capillari e vene. Questo permette di valutarne la distribuzione e di analizzare lo stato delle pareti vascolari, fornendo informazioni utili sulle condizioni della circolazione retinica.
Che decorso ha la malattia?
Il decorso della retinite pigmentosa è estremamente variabile da persona a persona, ma la malattia ha comunque un andamento progressivo e invalidante. Nella maggior parte dei casi, i disturbi visivi tendono ad accentuarsi nel tempo e il campo visivo si restringe gradualmente, fino a ridursi in alcuni casi in modo marcato. Con l’avanzare della patologia possono comparire anche altri sintomi, come una maggiore sensibilità alla luce, difficoltà nella percezione dei colori e lo sviluppo di cataratta. Purtroppo, nelle fasi più avanzate, l’evoluzione della malattia può condurre, in molti casi, a una grave compromissione della vista fino alla cecità.
Si può curare?

Attualmente la retinite pigmentosa non è considerata una malattia guaribile. In passato, molte speranze erano state riposte nella terapia iperbarica, nota anche come ossigenoterapia iperbarica(OTI), con l’obiettivo di rallentare o arrestare la progressione della patologia. Diversi studi hanno effettivamente evidenziato una risposta positiva a livello cellulare, confermata anche da esami strumentali come l’elettroretinogramma, che nei pazienti sottoposti al trattamento mostrava un aumento significativo dell’ampiezza del tracciato. Tuttavia, nonostante questi risultati incoraggianti, l’ossigenoterapia non si è dimostrata risolutiva e non interviene sulle cause profonde della malattia. Nonostante i progressi clinici concreti siano stati finora limitati, la ricerca scientifica è molto attiva e segue diversi filoni di studio. Attualmente le prospettive più promettenti riguardano la terapia genica e l’impiego di cellule staminali.
Le vitamine possono aiutare?
Negli ultimi anni è stata osservata una certa efficacia di terapie di supporto basate sull’assunzione di specifici integratori, in particolare nelle fasi iniziali della malattia, quando la visione centrale risulta ancora relativamente conservata. In questo contesto, alcuni studi hanno evidenziato un’associazione favorevole tra l’impiego della vitamina A e una gestione più efficace della retinite pigmentosa, soprattutto se inserita all’interno di un percorso di monitoraggio clinico. Benefici potenziali sono stati osservati anche con l’assunzione di acidi grassi omega‑3, antiossidanti e luteina, sostanze utili a sostenere la salute della retina e a contrastare i meccanismi di stress ossidativo. È fondamentale sottolineare che questi integratori devono essere assunti a dosaggi controllati e sempre sotto supervisione medica, poiché non rappresentano una cura, ma un possibile supporto volto a preservare più a lungo la funzione visiva residua.
Terapia genica
L’obiettivo della ricerca è individuare i geni responsabili della malattia per poter intervenire in modo mirato attraverso le moderne tecniche di ingegneria genetica. In questo ambito si punta, da un lato, a sostituire i geni alterati con copie sane e, dall’altro, soprattutto nelle forme a trasmissione autosomica dominante, a disattivare i geni che risultano dannosi. Per trasportare il materiale genetico corretto viene generalmente utilizzato un virus vettore, opportunamente modificato per renderlo innocuo. Questo virus, paragonabile a un “cavallo di Troia”, è in grado di introdurre all’interno delle cellule le sequenze di DNA necessarie alla correzione del difetto genetico. Attraverso iniezioni effettuate sotto la retina si tenta così di intervenire direttamente sulla causa della malattia. Questo approccio ha già mostrato risultati positivi soprattutto nei bambini affetti da un’altra patologia genetica retinica, l’amaurosi congenita di Leber. Nel caso della retinite pigmentosa, tuttavia, la presenza di un numero molto elevato di geni coinvolti rende il trattamento genetico più complesso e ancora oggetto di studio. Negli ultimi anni la ricerca scientifica ha compiuto importanti passi avanti nella comprensione delle cause genetiche della retinite pigmentosa e nello sviluppo di nuove terapie, in particolare nell’ambito della terapia genica. Nonostante questi progressi, al momento non esiste ancora un trattamento efficace valido per tutti i pazienti. Attualmente l’unica terapia genica approvata è quella con voretigene neparvovec, indicata però esclusivamente per le forme di retinite pigmentosa causate da una mutazione biallelica del gene RPE65. Parallelamente, la ricerca sta avanzando rapidamente anche per altre varianti della malattia: sono infatti in corso studi clinici in fase avanzata per alcune forme specifiche, come quella legata al cromosoma X associata al gene RPGR, oltre allo sviluppo di tecniche innovative di editing genetico, tra cui CRISPR-Cas9, che aprono nuove prospettive per il futuro.
Cellule staminali
La ricerca sulle cellule staminali applicata alla retinite pigmentosa si trova attualmente in una fase di rapido sviluppo. Sono in corso studi clinici di fase iniziale, mirati principalmente a valutare la sicurezza e l’efficacia di questi approcci terapeutici. Le cellule staminali vengono studiate con due obiettivi principali: da un lato, sostituire i fotorecettori che sono andati perduti a causa della degenerazione della retina; dall’altro, fornire un sostegno alle cellule retiniche ancora vitali, rallentando il processo degenerativo attraverso un effetto protettivo. In particolare, si stanno organizzando studi che utilizzano cellule staminali progenitrici della retina, somministrate mediante iniezione nella cavità vitreale, soprattutto nei casi di retinite pigmentosa in fase avanzata. Parallelamente, l’attenzione dei ricercatori si sta concentrando anche su altre tipologie cellulari, come le cellule staminali neurali e le cellule staminali mesenchimali derivate dalla gelatina di Wharton del cordone ombelicale. Queste ultime si sono dimostrate capaci di rilasciare sostanze protettive in grado di ridurre l’infiammazione e limitare la morte delle cellule retiniche. Un altro ambito di grande interesse è rappresentato dalle cellule staminali pluripotenti indotte, ottenute a partire da cellule adulte. Queste cellule hanno mostrato, nei modelli sperimentali, la capacità di differenziarsi in cellule retiniche funzionali senza evidenziare un rischio significativo di formazione tumorale, aprendo prospettive potenzialmente importanti per il futuro. Nonostante i risultati preliminari siano incoraggianti, la terapia con cellule staminali non è ancora considerata un trattamento standard per la retinite pigmentosa. Restano infatti aperte diverse sfide, in particolare quelle legate alla sicurezza nel lungo periodo e alla reale capacità delle nuove cellule di integrarsi in modo efficace e funzionale con il tessuto retinico esistente.
Trapianto di retina
Attualmente non esiste ancora un vero e proprio trapianto biologico della retina. Tuttavia, la ricerca sta esplorando approcci innovativi che potrebbero aprire nuove prospettive terapeutiche. Tra questi vi sono i sistemi di retina artificiale, basati su microchip in grado di sostituire parzialmente la funzione dei fotorecettori danneggiati. Questi dispositivi sono già utilizzati da alcuni anni e le sperimentazioni hanno dimostrato la capacità di riconoscere oggetti o contorni, migliorando in parte l’autonomia dei pazienti, anche se i risultati ottenuti non sono sempre pienamente soddisfacenti. Un’altra strategia emergente è rappresentata dalla cosiddetta “retina liquida”, un prototipo già testato con risultati promettenti, che prevede l’iniezione di nanoparticelle capaci di comportarsi in modo simile ai fotorecettori naturali. Anche questo approccio è ancora in fase sperimentale, ma offre interessanti prospettive per il futuro trattamento delle forme più avanzate della malattia.
Immunologia
Si prefigge di verificare alcune teorie che ipotizzerebbero un’alterazione del sistema immunitario che potrebbe essere il principale fattore scatenante della malattia. Le ricerche più recenti in ambito immunologico hanno messo in evidenza il ruolo fondamentale dell’infiammazione nella progressione della retinite pigmentosa, aprendo nuove prospettive terapeutiche. Diversi studi indicano infatti che, nella retina in fase degenerativa, si attivano processi infiammatori che contribuiscono alla morte dei fotorecettori, indipendentemente dalla specifica mutazione genetica responsabile della malattia. La perdita dei fotorecettori innesca una risposta immunitaria locale che coinvolge le cellule gliali, normalmente deputate al supporto delle cellule nervose. Tuttavia, quando questa attivazione diventa cronica, le cellule gliali possono rilasciare sostanze potenzialmente dannose, contribuendo ad accelerare il processo degenerativo. In alcuni casi, il sistema immunitario può arrivare ad aggredire erroneamente le cellule retiniche già compromesse, aggravando ulteriormente il danno. Alla luce di queste evidenze, la ricerca attuale si sta concentrando sul possibile riutilizzo di farmaci già disponibili per contrastare la componente infiammatoria della malattia. In particolare, studi recenti suggeriscono che l’impiego di farmaci antinfiammatori come i glucocorticoidi, potrebbero rallentare la perdita della funzione visiva e favorire la sopravvivenza dei coni, indipendentemente dal gene coinvolto. L’obiettivo di questo approccio non è correggere direttamente la causa genetica della retinite pigmentosa, ma proteggere i fotorecettori ancora integri, contribuendo a rallentare la progressione della malattia e a preservare più a lungo la funzione visiva residua.
Il futuro dei trattamenti
Come già accennato in precedenza, le possibilità di cura della retinite pigmentosa sembrano essere sempre più legate allo sviluppo della terapia genica e all’impiego delle cellule staminali. In alcuni pazienti che hanno perso la vista a causa della malattia sono stati impiantati microchip nella retina. Quando l’intervento ha avuto esito positivo, questi dispositivi hanno consentito di recuperare una forma di visione molto limitata, ma sufficiente in alcuni casi a migliorare l’autonomia e l’orientamento nello spazio.
Accanto alle nuove tecnologie terapeutiche, ancora in fase sperimentale, sono oggi disponibili numerose soluzioni di tipo assistivo pensate per le persone ipovedenti, con l’obiettivo di migliorare la qualità della vita quotidiana. Molte di queste tecnologie sono integrate in dispositivi di uso comune, come computer, smartphone, tablet che offrono funzioni quali l’ingrandimento delle immagini, l’aumento del contrasto tra chiaro e scuro e la conversione dei testi in formato audio. Alcune applicazioni consentono inoltre di utilizzare la fotocamera dello smartphone per riconoscere e descrivere gli oggetti inquadrati, facilitando l’orientamento nello spazio e la mobilità. Quando la funzione visiva non è completamente compromessa, possono essere intrapresi specifici percorsi di riabilitazione visiva. Questi trattamenti non mirano a ripristinare la vista, ma a gestire in modo più efficace la progressione della malattia e a valorizzare al massimo la capacità visiva residua, sfruttando i fotorecettori ancora funzionanti.
Nelle forme più avanzate di retinite pigmentosa, assume inoltre un’importanza fondamentale il supporto psicologico. Affrontare una diagnosi di questo tipo, insieme alla progressiva perdita della funzione visiva, può avere un forte impatto emotivo e comportare un aumento significativo del rischio di sviluppare disturbi dell’umore, come stati depressivi. Per questo motivo, è spesso consigliato intraprendere un percorso di sostegno psicologico, che può includere colloqui individuali o il coinvolgimento della famiglia, con l’obiettivo di favorire l’adattamento alla malattia, ridurre il senso di isolamento e migliorare il benessere complessivo della persona.
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Leggi le condizioni generali di consultazione di questo sito
Ultima revisione scientifica: 14 maggio 2026
