









È una procedura chirurgica che consiste nell’asportazione del corpo vitreo (sostanza gelatinosa e trasparente che riempie la cavità oculare tra il cristallino e la retina e che costituisce indicativamente i 4/5 del volume del bulbo oculare) e nella sua sostituzione con un mezzo analogo che viene inserito all’interno dell’occhio e definito “sostituto vitreale” (aria, gas, olio di silicone).
La vitrectomia può essere indicata per trattare diverse patologie oculari, principalmente si effettua in caso di:
Sì, ne presenta alcuni: la vitrectomia è un intervento delicato di microchirurgia. Le complicanze principali sono eventuali distacchi di retina, sanguinamenti, opacizzazioni del cristallino e glaucoma. (Leggi il consenso informato della SOI).
 No, il corpo vitreo non si rigenera spontaneamente nell’immediato: nell’occhio operato può essere inserito temporaneamente un sostituto vitreale che funziona sia come mezzo diottrico (attraverso di esso passa la luce), sia come mezzo tamponante (mantiene la retina in sede). Alcuni sostituti vitreali devono essere rimossi obbligatoriamente dopo un certo periodo di tempo, ad esempio l’olio di silicone va asportato dopo circa 3-6 mesi perché altrimenti potrebbe provocare glaucoma o danni corneali; invece altri mezzi, come l’aria o il gas, si riassorbono spontaneamente e vengono progressivamente sostituiti dai normali fluidi organici presenti all’interno dell’occhio.
No, il corpo vitreo non si rigenera spontaneamente nell’immediato: nell’occhio operato può essere inserito temporaneamente un sostituto vitreale che funziona sia come mezzo diottrico (attraverso di esso passa la luce), sia come mezzo tamponante (mantiene la retina in sede). Alcuni sostituti vitreali devono essere rimossi obbligatoriamente dopo un certo periodo di tempo, ad esempio l’olio di silicone va asportato dopo circa 3-6 mesi perché altrimenti potrebbe provocare glaucoma o danni corneali; invece altri mezzi, come l’aria o il gas, si riassorbono spontaneamente e vengono progressivamente sostituiti dai normali fluidi organici presenti all’interno dell’occhio.
La procedura più diffusa è la vitrectomia a tre vie. Si inseriscono nell’occhio tre strumenti. Il primo è la sonda che inietta l’acqua (BSS, soluzione salina bilanciata) per mantenere costanti la pressione e il volume dell’occhio; il secondo è una fibra ottica che serve ad illuminare; infine, c’è il vitrectomo, uno strumento che taglia e contemporaneamente aspira il corpo vitreo. Da diversi anni l’intervento chirurgico è stato migliorato, può essere infatti realizzato mediante micro-incisioni che, non necessitando di punti di sutura, favoriscono un rapido recupero funzionale e provocano minore infiammazione oculare. Questo tipo di vitrectomia è denominata mini-invasiva da 27G. Con la lettera “G” = Gauge, si indica la misura del diametro degli aghi delle siringhe, più è grande il numero più piccolo è il diametro. La vitrectomia mininvasiva consente, dunque, un rapido ripristino delle funzioni dell’occhio e un ricovero di qualche ora per il paziente, che potrà tornare a casa in tempi brevi dopo l’intervento. Il giorno successivo è prevista la visita di controllo con l’oculista, dopodiché il paziente potrà riprendere gradualmente le sue attività normali; per quelle più faticose (quali palestra o sport), sarà necessario attendere qualche settimana. Ovviamente, verranno programmate altre visite di controllo, secondo le tempistiche indicate dall’oculista.
La vitrectomia è un intervento che presenta alcuni rischi: l’occhio potrebbe diventare più delicato. Resta però l’unica soluzione per risolvere alcune patologie oculari e si rende necessario per evitare complicanze più gravi.
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Leggi le condizioni generali di consultazione di questo sito
Pagina pubblicata il 3 giugno 2009. Ultimo aggiornamento: 13 giugno 2025.
Ultima revisione scientifica: 13 giugno 2025.
Tumore intraoculare più diffuso
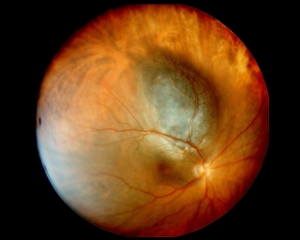 Il melanoma della coroide (tessuto vascolare tra la retina e la sclera) è il tumore maligno primitivo intraoculare più frequente nell’adulto, con un’incidenza pari a 6 casi su un milione di abitanti. Si presenta più comunemente nei soggetti di sesso maschile, con età compresa tra i 50 e i 60 anni; la razza bianca ha un rischio otto volte maggiore rispetto a quella nera.
Il melanoma della coroide (tessuto vascolare tra la retina e la sclera) è il tumore maligno primitivo intraoculare più frequente nell’adulto, con un’incidenza pari a 6 casi su un milione di abitanti. Si presenta più comunemente nei soggetti di sesso maschile, con età compresa tra i 50 e i 60 anni; la razza bianca ha un rischio otto volte maggiore rispetto a quella nera.
L’origine della neoplasia è dovuta a diverse cause ed è necessaria l’interazione di fattori genetici ed ambientali perché si sviluppi. Oltre all’età e alla razza i principali fattori di rischio sembrerebbero essere: colore chiaro della pelle, capelli biondi, iride chiara, esposizione al sole e presenza di nevi.
I melanomi insorgono nella maggioranza dei casi ex novo, mentre in una ridotta percentuale di casi si sviluppano a partire da un nevo. Alcuni nevi presentano un alto rischio di trasformazione maligna; di conseguenza richiedono un’accurata osservazione clinica. Il monitoraggio periodico avviene mediante valutazione oftalmoscopica ed attraverso il confronto nel tempo di fotografie della lesione (anche se biologicamente benigna). La frequenza dei controlli sarà inizialmente dettata dalle dimensioni e dalle caratteristiche cliniche del nevo: un nevo di spessore inferiore ai 2 mm, con drusen superficiali, assenza di pigmento arancio, a margini netti, asintomatico e senza alterazioni circostanti dell’epitelio pigmentato retinico, può essere controllato una volta all’anno. I nevi con spessore superiore a 2mm, segni di liquido sotto retinico, insorti in soggetti giovani, localizzati nella zona maculare o in prossimità del disco ottico, devono essere controllati ogni tre mesi o eventualmente sei. Segni di una trasformazione maligna sono facilmente valutabili, con l’esame oftalmoscopico ed ancor meglio con un’ecografia oculare.
Il melanoma della coroide si sviluppa generalmente senza sintomi specifici; tuttavia, qualora esso sia localizzato nella fovea (zona centrale della retina) o parafoveale (in prossimità del centro retinico) può determinare una riduzione dell’acuità visiva. Altri sintomi riferiti dai pazienti sono: fosfeni (lampi di luce a volte colorati), immagini distorte (metamorfopsie), alterazioni del campo visivo, dilatazione dei vasi sclerali, dolore (nelle fasi più avanzate).
Oggi il melanoma coroideale può essere diagnosticato con una precisione del 99,7% grazie alla notevole esperienza clinica che caratterizza i centri di oncologia oculare e alle moderne tecniche diagnostiche. L’esame oftalmoscopico indiretto e l’ecografia oculare sono le tecniche più importanti per poter giungere alla diagnosi. Con l’oftalmoscopia si possono individuare i seguenti aspetti clinici: forma, dimensione, colore, localizzazione, presenza di essudazione, emorragie, alterazioni a carico dell’epitelio pigmentato retinico. L’ecografia oculare con tecnica A-B scan consente, a sua volta, di definire altri aspetti della lesione come: la morfologia, la struttura interna e l’infiltrazione sclerale, al fine di determinare con precisione la natura del tumore, definirne le esatte dimensioni, valutarne il trattamento più appropriato.
Si può far ricorso alla fluorangiografia (FAG) che, al contrario dell’ecografia, non fornisce dati fondamentali ai fini diagnostici, ma risulta fondamentale per differenziare il melanoma da altre patologie, come ad esempio un’emorragia sottoretinica. Inoltre la FAG, insieme all’angiografia al verde di indocianina, può fornire utili indicazioni per differenziare il melanoma amelanotico da lesioni benigne ad alta vascolarizzazione (come l’emangioma della coroide).
Il nevo della coroide, l’ipertrofia dell’epitelio pigmentato retinico (EPR), le emorragie sottoretiniche, le metastasi coroideali, l’emangioma, l’osteoma ed il melanocitoma della coroide sono le condizioni di più frequente riscontro che devono essere differenziate dal melanoma coroideale. A tal fine occorre sempre eseguire un accurato esame clinico, ecografico ed, eventualmente, angiografico.
Il melanoma della coroide, metastatizza unicamente per via ematica, data l’assenza di vasi linfatici a livello del bulbo. Gli organi e le strutture che risultano maggiormente coinvolti da metastasi sono: il fegato (nella maggior parte dei casi), i polmoni, lo scheletro, la cute ed il sistema nervoso centrale.
 Oggi un trattamento di tipo conservativo è indicato nella maggior parte dei melanomi: in genere non è necessario asportare chirurgicamente l’occhio. Le tecniche conservative maggiormente utilizzate sono le seguenti:
Oggi un trattamento di tipo conservativo è indicato nella maggior parte dei melanomi: in genere non è necessario asportare chirurgicamente l’occhio. Le tecniche conservative maggiormente utilizzate sono le seguenti:
Esiste una tecnica di radioterapia ancora più potente, che sfrutta protoni accelerati per colpire le cellule tumorali: fa effetto fino a 14 mm di profondità (l’adroterapia). Tale metodica, pertanto, risulta particolarmente indicata in pazienti monocoli con tumori di spessore superiore a quello consentito per il trattamento con brachiterapia o con terapia sandwich.
Anche nei casi in cui le dimensioni della lesione, l’estensione extrasclerale, l’associazione a glaucoma secondario impongano un intervento demolitivo (enucleazione del bulbo oculare), le tecniche operatorie e i materiali oggi disponibili consentono di ripristinare un aspetto estetico assolutamente soddisfacente, con evidente beneficio psicologico. Un’adeguata programmazione dei controlli oftalmoscopici ed ecografici dopo il trattamento, con cadenza semestrale nei primi cinque anni e annuale nel periodo successivo, è parte integrante del programma terapeutico.
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Leggi le condizioni generali di consultazione di questo sito
Pagina pubblicata il 3 luglio 2008. Ultimo aggiornamento: 13 giugno 2025
Ultima revisione scientifica: 13 giugno 2025
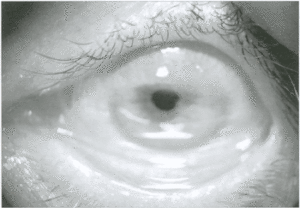 Si tratta di una grave malattia infettiva degli occhi, una tra le più antiche conosciute dal genere umano, provocata dal batterio Chlamydia Tracomatis. Nella diffusione della patologia occorre tenere in considerazione vari fattori:
Si tratta di una grave malattia infettiva degli occhi, una tra le più antiche conosciute dal genere umano, provocata dal batterio Chlamydia Tracomatis. Nella diffusione della patologia occorre tenere in considerazione vari fattori:
Il tracoma quasi sempre colpisce entrambi gli occhi. Il periodo di incubazione della malattia è tra i 5 e i 12 giorni, dopodiché compaiono i primi sintomi: occhi rossi, visione offuscata, palpebre gonfie, fastidio alla luce (fotofobia), lacrimazione, scolo nasale frequente. Negli stadi più avanzati della malattia i pazienti presentano: infezioni o infiammazioni ripetute, palpebre rivolte verso l’interno del bulbo (entropion), ciglia anch’esse rivolte all’interno (trichiasi), forte dolore oculare, visus ridotto e cecità, con formazione di cicatrici più o meno estese sulla cornea (nei casi più gravi si evidenzia un’opacizzazione completa della stessa e si forma un vero e proprio panno corneale).
Il tracoma è responsabile della cecità o della disabilità visiva di circa 1,9 milioni di persone e causa circa l’1,4% di tutta la cecità nel mondo [1]. Sulla base dei dati di aprile 2024, risulta che 103 milioni di persone vivono in aree in cui il tracoma è endemico e sono a rischio cecità; tali aree sono quella più povere e rurali dell’Africa, dell’America centrale e meridionale, dell’Asia, dell’Australia e del Medio Oriente. Nel complesso, l’Africa rimane il continente più colpito e quello in cui si concentrano gli sforzi di controllo più intensi. Ad ottobre 2024, 21 paesi (Benin, Cambogia, Cina, Gambia, Repubblica islamica dell’Iran, Repubblica democratica popolare del Laos, Ghana, India, Iraq, Malawi, Mali, Messico, Marocco, Myanmar, Nepal, Oman, Pakistan, Arabia Saudita, Togo, Vanuatu e Vietnam) sono stati convalidati dall’OMS come paesi che hanno eliminato il tracoma come problema di salute pubblica. Nel 2023, 130.746 persone hanno ricevuto un trattamento chirurgico per lo stadio avanzato della malattia e 32,9 milioni di persone sono state curate con antibiotici. La copertura antibiotica globale nel 2023 è stata del 29%.
I programmi internazionali di prevenzione e di lotta alla malattia, portati avanti in particolare dall’OMS e dalla IAPB (Sezione italiana), negli anni stanno dando risultati positivi.
In aree dove è diffusa, l’infezione da tracoma colpisce quasi tutti i bambini nei primi due anni di vita. I piccoli malati sono la principale riserva d’infezione nella comunità, soprattutto per le donne che vi stanno a più stretto contatto, giustificando l’incidenza due o tre volte più alta della forma più grave di tracoma nel sesso femminile rispetto a quello maschile. I bambini ospitano il batterio nei tratti superiori dell’apparato respiratorio e gastrointestinale, cosicché la trasmissione può avvenire anche attraverso la propagazione di goccioline respiratorie oppure contaminazione per mezzo delle feci. Con la crescita dei bambini diminuisce la prevalenza e la gravità della malattia attiva. Verso i 15-20 anni inizia la fase cronica cicatriziale della patologia.
Il tracoma è molto probabilmente dovuto a una risposta di carattere immunologico in conseguenza di ripetute e continue esposizioni al batterio Chlamydia Tracomatis. Un tracoma infettivo acuto è caratterizzato da una congiuntivite non purulenta in entrambi gli occhi e generalmente si evidenzia gonfiore a livello delle ghiandole linfatiche del collo e davanti all’orecchio.
L’Organizzazione Mondiale della Sanità (OMS) ha identificato cinque stadi nello sviluppo del tracoma:
Durante la visita medica l’oculista, in prima istanza, prende in esame l’anamnesi del paziente ponendo specifiche domande per verificare se di recente sia stato in aree affette dal tracoma. Poi, procede con l’ esame obiettivo alla lampada a fessura per individuare l’eventuale presenza di cicatrici all’interno della palpebra, le condizioni della cornea e la presenza di possibili follicoli sulla congiuntiva. Per effettuare una diagnosi precisa lo specialista potrà prelevare un campione dall’occhio del paziente per inviarlo al laboratorio. Per individuare la presenza del batterio, si potranno eseguire i seguenti test: analisi PCR per il DNA del batterio, esame culturale, test di immunofluorescenza diretta.

Nelle forme acute sporadiche il trattamento con antibiotici per via sistemica è sufficiente. Il trattamento del tracoma endemico rappresenta, invece, una sfida particolare. VISION 2020 – un progetto mondiale per eliminare la cecità evitabile portato avanti dalla IAPB e inizialmente dall’OMS – comprende un programma specifico per l’eliminazione globale del tracoma attuando la cosiddetta strategia “SAFE“. L’acronimo S.A.F.E. rappresenta le quattro componenti chiave di questa strategia:
È stato fissato il 2030 come nuova data per riuscire ad ottenere l’eliminazione globale del tracoma (insieme ad altre malattie).
[1] Fonte: https://www.who.int/news-room/fact-sheets/detail/trachoma
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Leggi le condizioni generali di consultazione di questo sito
Pagina pubblicata il 27 aprile 2007. Ultimo aggiornamento: 25 marzo 2025
Ultima revisione scientifica: 25 marzo 2025
Il campo visivo è quella porzione di spazio che l’occhio è in grado di percepire, quando viene osservato un punto fisso. Occorre fare una distinzione tra campo visivo monoculare e campo visivo binoculare: il primo rappresenta ciò che è visibile con un solo occhio, mentre il secondo si riferisce ad entrambi gli occhi. Di norma, il campo visivo viene suddiviso in quattro quadranti, ottenuti tramite l’intersezione di due assi perpendicolari in un punto, detto punto di fissazione e corrispondente alla fovea retinica. La fovea è la regione centrale della retina in cui si ha la massima acutezza visiva.
Lo studio del campo visivo può essere effettuato mediante perimetria manuale o automatica; quest’ultima attualmente è sicuramente la più diffusa e si avvale di macchinari automatizzati che rendono più semplice l’analisi dei risultati grazie alla presenza di dispositivi hardware e software. Nella perimetria automatizzata, il campo visivo viene valutato qualitativamente e quantitativamente. Il perimetro automatico consente di testare un numero maggiore di punti rispetto a quello manuale. In particolare, grazie a degli algoritmi numerici è possibile determinare la distribuzione del difetto e la sensibilità della retina, dati che vengono poi comparati con i risultati della popolazione “normale” (cioè senza deficit del campo visivo). Attualmente, alla ‘’perimetria’’ viene associato il termine ‘’campimetria’’, in quanto si utilizzano moderne apparecchiature computerizzate (i campimetri) che elaborano vere e proprie mappe del campo visivo.
Per il funzionamento del campo esistono due metodi: il “cinetico” e lo “statico”. Tali metodi si differenziano a seconda di come vengono proiettate le luci su uno schermo. Il metodo cinetico utilizza luci che si muovono (stimoli luminosi) dall’esterno all’interno variando al contempo la loro intensità luminosa. Col metodo statico si utilizzano, invece, luci fisse; ma anche in questo caso varia la loro luminosità. Per l’esecuzione del campo visivo, il paziente è seduto di fronte ad una piccola cupola concava con un target al centro e gli viene chiesto di focalizzarsi su di esso. L’occhio non in esame viene coperto. Il computer dà l’input di stimolo luminoso alla cupola e il paziente preme un pulsante ogni volta che ne riconosce uno. Sarà il computer stesso a fornire una mappatura automatica e a calcolare il campo visivo del paziente. Per valutare l’attendibilità dei risultati del campo visivo, l’oculista deve controllare appositi parametri (definiti appunto come indici di affidabilità):
perdite di fissazione, esprime il numero di risposte affermative alla presentazione di stimoli presenti nella macchia cieca, il che indica che in quel momento il paziente non fissava la mira centrale. Un numero alto di perdite di fissazione, può indicare una scarsa comprensione da parte del paziente di come deve essere eseguito il test, affaticamento, agitazione, scarsa collaborazione.
L’Humphrey Field Analyzer (HFA) rappresenta il gold standard di riferimento per la perimetria e, in particolare, per la diagnosi del glaucoma.
Il metodo di Zingirian-Gandolfo permette un’analisi completa del campo visivo binoculare, con calcolo della minorazione della visione periferica (fino all’ipovisione o alla cecità). Consente di effettuare una valutazione ottimale a livello funzionale, dato importante per quantificare il danno visivo periferico e, di conseguenza, le difficoltà avvertite dal paziente nello svolgimento delle normali attività quotidiane. Il campo visivo percentuale di Zingirian-Gandolfo ha tantissimi vantaggi:
Il test si fa con entrambi gli occhi aperti, facendo fissare il paziente con l’occhio migliore. Ha una estensione orizzontale complessiva di circa 120°.Con tale metodo vengono esplorati 100 punti, dei quali 60 si trovano nella zona bassa del campo visivo (emicampo inferiore) e i rimanenti 40 in quella superiore. Inoltre 64 punti sono collocati nel campo visivo paracentrale (5°-30°) e solo 36 in quello periferico (30°-60°).
Il campo visivo permette di quantificare e rilevare le perdite assolute e relative di sensibilità retinica, evidenziando gli eventuali difetti periferici o centrali. Quindi è estremamente utile per valutare non solo l’esordio, ma anche la progressione di una malattia tanto invalidante quale il glaucoma.
Viene, inoltre, utilizzato per studiare le alterazioni campimetriche che si riscontrano nelle patologie neurologiche (neuriti ottiche, edema della papilla) e nelle affezioni cerebro-vascolari (ischemia del nervo ottico). L’architettura delle zone cieche del campo visivo (scotomatose) segue la disposizione delle fibre nervose, quindi, la valutazione di queste zone ci consente di risalire alla patologia in atto.
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Leggi le condizioni generali di consultazione di questo sito
Pagina pubblicata il 29 gennaio 2009. Ultimo aggiornamento: 11 febbraio 2019.
Ultima revisione scientifica: 13 marzo 2025
Il glaucoma è una patologia oculare cronica che evolve lentamente ma in modo irreversibile, spesso senza dare segnali evidenti nelle prime fasi. La malattia provoca un danno alle cellule nervose della retina e alla testa del nervo ottico (con conseguente compromissione del campo visivo) ed è spesso correlata a una pressione intraoculare elevata. Nel glaucoma, considerata ormai una patologia neurodegenerativa a tutti gli effetti, si verifica la morte di una specifica popolazione di cellule della retina, le cellule ganglionari. In un occhio sano, le cellule ganglionari svolgono un ruolo fondamentale: ricevono le informazioni luminose dai fotorecettori — che catturano le immagini — e le trasmettono al cervello attraverso gli assoni, che insieme formano il nervo ottico. Nel glaucoma, queste cellule vanno incontro a morte progressiva, interrompendo la comunicazione tra occhio e cervello e compromettendo così la vista. I danni provocati dal glaucoma possono sfociare in ipovisione o disabilità visiva, ma la buona notizia è che, se la malattia viene scoperta e trattata in tempo, è possibile fermarne la progressione. Per questo è fondamentale sottoporsi a controlli regolari e non trascurare eventuali segnali di alterazioni della vista: il glaucoma può essere silenzioso, ma una diagnosi precoce può davvero fare la differenza.
Epidemiologia
Il glaucoma rappresenta una delle principali cause di cecità nel mondo. Si stima che, nelle persone tra i 40 e gli 80 anni, la prevalenza del glaucoma ad angolo aperto sia intorno al 3,5%, mentre il glaucoma ad angolo chiuso colpisca circa lo 0,5% della popolazione. Secondo l’Organizzazione Mondiale della Sanità, nel mondo ci sono oltre 76 milioni di persone affette da questa malattia [1] e si prevede che il numero continuerà a crescere nei prossimi decenni a causa dell’invecchiamento della popolazione. Si tratta della seconda causa di cecità a livello planetario dopo la cataratta, ma è la prima a carattere irreversibile [2]. In Italia si stima che la malattia colpisca circa un milione di persone, ma quasi la metà non ne sarebbe a conoscenza perché non si sottopone regolarmente a controlli oculistici completi e a esami specifici come il fondo oculare, la tonometria e il campo visivo. Anche l’etnia gioca un ruolo nella prevalenza della patologia: il glaucoma ad angolo aperto è più diffuso nei soggetti di etnia nera, mentre il glaucoma ad angolo chiuso è più frequente nelle popolazioni dell’Asia orientale.
Altri fattori di rischio per lo sviluppo di glaucoma primario da chiusura d’angolo sono: l’età avanzata, la familiarità, il sesso femminile e l’ipermetropia.
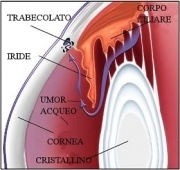
All’interno dell’occhio è presente un liquido chiamato umore acqueo, che viene continuamente prodotto e riassorbito. In questo senso, l’occhio può essere paragonato a un piccolo serbatoio con un rubinetto e una via di deflusso sempre aperti. Quando il deflusso dell’umore acqueo è ostruito, la pressione all’interno dell’occhio aumenta, determinando un rialzo della pressione intraoculare. Se questo aumento persiste nel tempo, il bulbo oculare può subire danni, in particolare a livello della testa del nervo ottico — la cosiddetta papilla ottica, situata nella parte centrale della retina. Poiché i danni al nervo ottico sono irreversibili, è fondamentale intervenire precocemente con una terapia adeguata.
COME SI PRODUCE IL DANNO ALLA VISTA?
La percezione visiva non riguarda soltanto l’oggetto che fissiamo, ma tutto ciò che lo circonda; l’insieme di ciò che riusciamo a vedere in un dato momento costituisce il cosiddetto campo visivo. Le immagini catturate dagli occhi vengono trasmesse dalla retina al cervello tramite il nervo ottico, che può essere paragonato a un cavo elettrico composto da milioni di fili, ciascuno dei quali trasporta informazioni relative a una piccola porzione del campo visivo. Il cervello, ricevendo questi segnali bioelettrici, riesce a ricostruire l’immagine nella sua interezza, permettendoci di percepire il mondo intorno a noi in modo coerente. Tuttavia, un aumento della pressione oculare può danneggiare irreparabilmente i neuroni responsabili di questo trasferimento di informazioni, come se i fili del “cavo” si logorassero. In particolare, la morte delle cellule ganglionari retiniche interrompe la comunicazione tra i fotorecettori della retina e il cervello, provocando una perdita di campo visivo. La gravità di questa perdita dipende dall’entità del danno: più cellule vengono compromesse, più importante sarà la riduzione della nostra capacità visiva. Pertanto, la salute delle cellule ganglionari, risulta fondamentale per mantenere una visione completa e nitida del mondo circostante. Esiste infatti una corrispondenza topografica per cui la perdita di cellule ganglionari in una specifica area della retina corrisponde alla perdita di una parte di campo visivo. Nel glaucoma, il danno inizia generalmente dalle fibre che trasportano le informazioni provenienti dalla periferia del campo visivo. In questa fase, la persona continua a vedere chiaramente l’oggetto su cui fissa lo sguardo, ma non si accorge che la visione laterale si sta progressivamente riducendo. Con l’avanzare della malattia, anche le fibre provenienti dalla macula, la zona centrale della retina responsabile della visione dettagliata, vengono danneggiate. Questo comporta una significativa riduzione dell’acuità visiva. Come avviene in molte patologie neurodegenerative, il danno causato dal glaucoma non è reversibile: se la malattia non viene trattata tempestivamente e con successo, può portare a una perdita della vista parziale o addirittura totale.
SINTOMI DELLA PRESSIONE OCULARE ALTA
Nelle varie forme di glaucoma, ad eccezione di quello acuto, la malattia insorge e si sviluppa senza che il paziente avverta sintomi particolari. Quando il soggetto si rende conto di non vedere bene nella parte periferica del proprio campo visivo, purtroppo i danni a carico delle fibre del nervo ottico sono già presenti. La visione centrale di solito è ben conservata, il visus può essere anche pari a 10/10, ma il paziente ha evidenti difficoltà nello svolgimento di azioni che sfruttano la parte periferica del campo visivo (scendere le scale, guidare, attraversare la strada).
I sintomi che possono comunque presentarsi in caso di pressione oculare alta sono:
Con il termine glaucoma si indica un gruppo di patologie a diversa eziologia e con decorso clinico eterogeneo. A scopo meramente classificativo, si possono distinguere i glaucomi in due grandi famiglie: i glaucomi ad angolo aperto e i glaucomi da chiusura d’angolo, i quali differiscono significativamente per meccanismo patogenetico e per decorso clinico.
Ø Glaucoma primario ad angolo aperto (POAG)
Rappresenta la forma più comune di glaucoma nei paesi occidentali, è caratterizzata da un angolo irido-corneale di ampiezza normale. I principali fattori di rischio sono rappresentati da una pressione intraoculare (IOP) elevata e dall’età avanzata. La patogenesi del danno da POAG è, ad oggi, ancora incerta: si ritiene che la pressione intraoculare elevata, in concomitanza ad alterazioni vascolari, abbia un ruolo nell’induzione della morte delle cellule ganglionari retiniche e dei loro assoni, probabilmente ostacolando il normale trasporto assonale che avviene in queste cellule. L’aumento progressivo della pressione intraoculare sembra essere invece determinato da un aumento della resistenza a livello del trabecolato, ossia la parte dell’occhio deputata al filtraggio e allo “scarico” dell’umor acqueo: l’alterazione del trabecolato (dovuta all’età, allo stress ossidativo o ad altri fattori sconosciuti) comporta quindi una maggior difficoltà a drenare il liquido presente nell’occhio con conseguente incremento della pressione oculare . Una distinzione piuttosto arbitraria è stata proposta per dividere i POAG in base al livello di pressione intraoculare, esistono infatti i cosiddetti POAG a pressione elevata e i POAG a pressione normale: nel caso di questi ultimi i pazienti presentano livelli di pressione intraoculare considerati “normali” (di solito < 20 mmHg), oltre ad avere delle caratteristiche cliniche peculiari. Si ritiene che nel caso dei POAG a pressione normale, fattori di rischio diversi dalla pressione intraoculare elevata abbiano un particolare peso specifico nel determinare l’inizio e il peggioramento della malattia; tuttavia i principi di trattamento rimangono gli stessi (vedi sezione “terapia”).
· ØPOAG sospetto ed ipertensione oculare
Può sembrare paradossale, ma non sempre le visite e gli esami strumentali riescono a fornire una chiara indicazione sul fatto che il paziente possa essere sano o affetto da glaucoma. Una percentuale non trascurabile di pazienti infatti, soprattutto in fase iniziale di malattia, vengono considerati con sospetto glaucoma. Questi soggetti presentano uno o più segni clinici suggestivi per glaucoma, ad esempio un aspetto della papilla ottica o un esame del campo visivo più o meno alterati, una pressione oculare borderline, misurazioni OCT che deviano dai valori normali; ma nessuno di questi esami è alterato in modo significativo o comunque in maniera concorde rispetto agli altri. Soltanto il tempo e la ripetizione periodica dei suddetti esami di solito riescono a chiarire il quadro clinico del paziente. Sarà l’oculista a stabilire se, nel frattempo, il paziente dovrà eseguire una terapia o meno, in base ai dati a disposizione e ai fattori di rischio di ciascun individuo. Soggetti con pressione oculare superiore ai valori considerati normali (> 21 mmHg) ma con nervo ottico sano, campo visivo normale, anatomia non suggestiva di possibile chiusura angolare e nessun altro fattore di rischio per glaucoma, sono definiti ipertesi oculari. Avere una pressione oculare elevata NON significa avere il glaucoma. Anche in questo caso, sarà l’oculista a valutare la necessità di una terapia e a programmare gli esami di follow-up, in base al rischio di conversione da ipertensione oculare a glaucoma.
Ø Glaucomi secondari ad angolo aperto
Costituiscono un gruppo di patologie accomunate dal riscontro di un angolo irido-corneale aperto, dallo sviluppo di un danno al nervo ottico di tipo glaucomatoso e, cosa che le differenzia dai glaucomi primari, da una causa chiaramente responsabile dello sviluppo della malattia. Di seguito, verranno brevemente trattati soltanto i glaucomi secondari più frequenti.
È il più frequente, ed è associato alla Sindrome Pseudoesfoliativa (detta anche PEX). È una situazione clinica caratterizzata dalla produzione di materiale furfuraceo da parte della superficie del cristallino che esfoliandosi intasa le vie di deflusso dell’umore acqueo (trabecolato) depositandosi a livello dell’angolo irido-corneale. Pertanto, nel tempo si determina un aumento della pressione intraoculare (IOP) e, di conseguenza, un glaucoma. Inoltre il materiale furfuraceo può depositarsi a livello dell’apparato di sospensione del cristallino al corpo ciliare (fibre zonulari), creando un indebolimento di tali fibre e una dislocazione del cristallino (sublussazione).Con il termine glaucoma si indica un gruppo di patologie a diversa eziologia e con decorso clinico eterogeneo. A scopo meramente classificativo, si possono distinguere i glaucomi in due grandi famiglie: i glaucomi ad angolo aperto e i glaucomi da chiusura d’angolo, i quali differiscono significativamente per meccanismo patogenetico e per decorso clinico
Ø Glaucoma primario ad angolo aperto (POAG)
Rappresenta la forma più comune di glaucoma nei paesi occidentali, è caratterizzata da un angolo irido-corneale di ampiezza normale. I principali fattori di rischio sono rappresentati da una pressione intraoculare (IOP) elevata e dall’età avanzata. La patogenesi del danno da POAG è, ad oggi, ancora incerta: si ritiene che la pressione intraoculare elevata, in concomitanza ad alterazioni vascolari, abbia un ruolo nell’induzione della morte delle cellule ganglionari retiniche e dei loro assoni, probabilmente ostacolando il normale trasporto assonale che avviene in queste cellule. L’aumento progressivo della pressione intraoculare sembra essere invece determinato da un aumento della resistenza a livello del trabecolato, ossia la parte dell’occhio deputata al filtraggio e allo “scarico” dell’umor acqueo: l’alterazione del trabecolato (dovuta all’età, allo stress ossidativo o ad altri fattori sconosciuti) comporta quindi una maggior difficoltà a drenare il liquido presente nell’occhio con conseguente incremento della pressione oculare . Una distinzione piuttosto arbitraria è stata proposta per dividere i POAG in base al livello di pressione intraoculare, esistono infatti i cosiddetti POAG a pressione elevata e i POAG a pressione normale: nel caso di questi ultimi i pazienti presentano livelli di pressione intraoculare considerati “normali” (di solito < 20 mmHg), oltre ad avere delle caratteristiche cliniche peculiari. Si ritiene che nel caso dei POAG a pressione normale, fattori di rischio diversi dalla pressione intraoculare elevata abbiano un particolare peso specifico nel determinare l’inizio e il peggioramento della malattia; tuttavia i principi di trattamento rimangono gli stessi (vedi sezione “terapia”).
Come si presenta la PEX?
Può essere evidenziata dall’oculista durante l’esame obiettivo con la lampada a fessura (meglio ancora se con la pupilla dilatata). Si potrà ben vedere il deposito del materiale furfuraceo sulla faccia anteriore del cristallino, distribuito in maniera circolare in campo pupillare e in periferia, assente laddove l’iride compie un movimento di sfregamento.
Quali sono gli aspetti caratteristici del glaucoma pseudoesfoliativo?
Di solito colpisce entrambi gli occhi (ossia è bilaterale) e spesso le alterazioni riscontrate sono asimmetriche nei due occhi; si evidenziano valori della pressione oculare piuttosto elevati, che possono subire importanti fluttuazioni nell’ambito delle 24 ore; ha un’evoluzione abbastanza rapida; c’è maggiore predisposizione allo sviluppo della cataratta; è presente una maggiore percentuale di complicanze durante e dopo gli interventi di cataratta; la pupilla si dilata con più difficoltà.
Come si cura il glaucoma pseudoesfoliativo?
Il trattamento di prima scelta è quello di tipo farmacologico, con l’instillazione di colliri che servono ad abbassare la pressione oculare (ipotonizzanti). La terapia deve essere tempestiva ed efficace per cui, se non si ottengono buoni risultati, all’inizio è bene ricorrere a più somministrazioni quotidiane (eventualmente con farmaci diversi). Il trattamento laser (trabeculoplastica) riceve spesso una buona risposta anche se non duratura, nel qual caso occorre ricorrere all’approccio chirurgico tramite l’intervento di trabeculectomia. È bene eseguire la facoemulsificazione quando è presente la cataratta (anche se in fase iniziale) e spesso anche quando il cristallino è ancora trasparente. Tale intervento, infatti, può indurre un significativo abbassamento della pressione intraoculare.
Inoltre, se effettuato precocemente è meno rischioso, in quanto la pupilla ha ancora una buona capacità dilatativa, c’è un basso rischio di dislocazione del cristallino nel vitreo durante l’operazione e si semplifica, infine, l’eventuale intervento di trabeculectomia (più facile da eseguire se il cristallino è già stato asportato in precedenza). Vale la pena, infine, ricordare l’importanza di controlli regolari per chi avesse una sindrome pseudoesfoliativa, ma non presenta ancora glaucoma. La misurazione periodica del tono, con esecuzione del campo visivo e della pachimetria nei casi sospetti (pressione oculare lievemente oltre i limiti della norma), aiuta a fare una diagnosi precoce e, quindi, a iniziare la giusta terapia prima che compaiano gravi danni oculari.
È associato alla sindrome da dispersione di pigmento. In questi pazienti, una particolare conformazione di alcune strutture intraoculari determina la liberazione di pigmento nell’umor acqueo, con conseguente alterazione della funzione trabecolare e incremento della pressione intraoculare. È più frequente nei maschi over 30. Alcune caratteristiche tipiche di questa condizione rendono generalmente agevole il riconoscimento di tale sindrome nei pazienti affetti da parte dell’oculista, che dovrà valutare la presenza o meno di glaucoma e la necessità o meno di una terapia. Le terapie e gli esami di follow-up sono gli stessi del POAG.
Altri glaucomi secondari ad angolo aperto:
Ø Glaucomi da chiusura angolare
Eterogeneo gruppo di condizioni patologiche che condividono, come principale responsabile dell’incremento della pressione intraoculare, e successivamente del danno glaucomatoso, l’apposizione della periferia iridea al trabecolato, la cosiddetta chiusura angolare. In questa condizione, il deflusso dell’umor acqueo attraverso il trabecolato viene ostacolato meccanicamente e si possono verificare quadri clinici più o meno severi ed eclatanti, dal glaucoma acuto alla chiusura angolare intermittente. I meccanismi patogenetici con i quali si innesca la chiusura angolare sono molteplici, il più frequente è il cosiddetto blocco pupillare (circa 2/3 dei casi), ossia il blocco del passaggio dell’umor acqueo attraverso il forame pupillare; altri meccanismi di induzione della chiusura angolare sono riconducibili ad alterazioni dell’anatomia dell’iride (iris plateau), ad alterazioni a livello del cristallino, ad alterazioni nella porzione posteriore dell’occhio, a somministrazione di farmaci o interventi di chirurgia oculare. I glaucomi da chiusura d’angolo sono meno frequenti nel mondo occidentale (vedi fattori di rischio), ma abbastanza frequenti per costituire una minaccia non trascurabile soprattutto in pazienti con determinati fattori di rischio, come l’ipermetropia. Il rischio di glaucoma da chiusura d’angolo deve essere sempre valutato dall’oculista durante le visite oculistiche di follow-up, perché in presenza di condizioni predisponenti, l’esecuzione di determinate procedure laser o chirurgiche (rispettivamente l’iridotomia YAG laser e l’intervento di cataratta a scopo idrodinamico) possono abbattere o addirittura eliminare l’eventualità futura di una chiusura angolare.
Ø Glaucoma congenito
In questa forma della patologia oculare il sistema di drenaggio è “cattivo” sin dalla nascita. Per questo motivo si verifica un aumento di pressione intraoculare. Il bambino presenta fastidio alla luce (fotofobia) e lacrimazione eccessiva. L’aumento pressorio può causare un aumento delle dimensioni dell’occhio (nei piccoli le pareti oculari sono meno resistenti) e la cornea può divenire opaca. Ogni sintomo sospetto deve indurre i genitori ad andare dall’oculista per effettuare una visita di controllo. Questo tipo di glaucoma è però raro: colpisce un neonato ogni diecimila.
L’unico modo per individuare precocemente il glaucoma è sottoporsi regolarmente a una visita oculistica completa. Prima di iniziare l’esame vero e proprio, l’oculista raccoglie i dati anamnestici del paziente, cercando di comprendere eventuali disturbi già presenti e verificando se in famiglia ci siano casi di glaucoma, poiché l’ereditarietà rappresenta un fattore importante nello sviluppo della malattia. Di seguito verranno elencati i principali esami eseguiti nella diagnosi e nel follow-up dei pazienti con glaucoma o con sospetto glaucoma.
Visita oculistica
Permette di valutare le diverse strutture dell’occhio coinvolte nello sviluppo e nella progressione della malattia. Durante l’esame, l’oculista può verificare, tra le altre cose, l’ampiezza della camera anteriore, importante nel glaucoma ad angolo chiuso, individuare eventuali cause di glaucoma secondario come la sindrome pseudoesfoliativa, la sindrome da dispersione di pigmento o la neovascolarizzazione, e osservare l’aspetto della testa del nervo ottico per identificare l’eventuale presenza di un danno da glaucoma più o meno avanzato. La visita oculistica deve comprendere la valutazione del fondo dell’occhio, preferibilmente in midriasi (mediante dilatazione della pupilla). In base alla gravità della malattia, alla rapidità di progressione della stessa, al tempo trascorso dalla diagnosi e a numerosi altri fattori, l’oculista deciderà quando eseguire la visita e/o gli esami di follow-up, generalmente in un tempo compreso tra i 3 e i 12 mesi. È importante ricordare come il glaucoma sia una malattia cronica, da cui non si può guarire; è necessario dunque effettuare delle visite e degli esami per tutta la vita, in modo tale da accorgersi per tempo di eventuali peggioramenti della malattia e prendere le giuste contromisure terapeutiche.
Misurazione della pressione oculare
La tonometria, uno degli esami fondamentali durante la visita oculistica, è un elemento chiave nella valutazione dei pazienti con glaucoma e nel monitoraggio dell’efficacia della terapia ipotonizzante. Esistono diversi strumenti per misurare la pressione intraoculare, ma il riferimento standard è il tonometro ad applanazione di Goldmann. Questo esame non è né doloroso né invasivo: lo strumento sfiora delicatamente la superficie dell’occhio dopo l’applicazione di un collirio anestetico e di un colorante (fluoresceina), permettendo di ottenere una misurazione accurata della pressione oculare. Oltre al tonometro ad applanazione, esistono altri strumenti per misurare la pressione intraoculare, come il tonometro a soffio e quello a rimbalzo. Questi dispositivi non sono invasivi e possono essere utilizzati senza l’applicazione del collirio anestetico o del colorante. La misurazione della pressione oculare può essere effettuata una sola volta nell’arco della giornata oppure più volte, a seconda delle necessità cliniche e della valutazione dell’oculista, ad esempio per eseguire una curva tonometrica e monitorare meglio le variazioni della pressione durante il giorno.
Esame del campo visivo
Esame strumentale che non può mancare nell’inquadramento diagnostico e nel monitoraggio dei pazienti con glaucoma o con sospetto glaucoma. Il campo visivo è la porzione di spazio che può essere percepita da un occhio ed il danno causato dal glaucoma consiste nella riduzione progressiva dell’estensione del campo visivo stesso. Gli strumenti a nostra disposizione (detti perimetri), riescono a rilevare danni al campo visivo molto precocemente, prima che il paziente stesso sviluppi dei sintomi. L’esame non è invasivo e consiste nel proiettare degli stimoli luminosi sullo sfondo chiaro del perimetro, la percezione di tali stimoli da parte del paziente dovrà essere confermata premendo un pulsante. Eseguire un campo visivo in modo tale che sia attendibile richiede un certo grado di collaborazione e concentrazione da parte del paziente, possono quindi essere necessari diversi esami prima che il paziente acquisisca l’esperienza necessaria ad eseguirlo in maniera corretta. Così come la visita oculistica, anche il campo visivo deve essere ripetuto a intervalli regolari, a discrezione dell’oculista.
OCT
Acronimo di tomografia a coerenza ottica, l’OCT è un esame strumentale non invasivo, che consente di ottenere immagini tomografiche ad alta risoluzione delle strutture coinvolte nel danno glaucomatoso. L’OCT costituisce un importante ausilio per l’oculista nella valutazione della morfologia di tali strutture ed è complementare alla visita oculistica con esame del fondo dell’occhio. Gli strumenti attualmente a disposizione riescono a fornire delle immagini a risoluzione e dettaglio molto elevati. L’OCT ha ormai affiancato la visita oculistica e il campo visivo nella diagnosi e nel follow up dei pazienti con glaucoma conclamato o sospetto e la periodicità con cui ripetere l’esame è stabilita dall’oculista.
Pachimetria
È la misurazione dello spessore della cornea (la porzione più anteriore dell’occhio) con strumenti che richiedono o meno il contatto con la superficie dell’occhio. Viene generalmente eseguita una sola volta e dovrebbe far parte degli esami di inquadramento di tutti i pazienti glaucomatosi o con sospetto glaucoma. Avere una cornea sottile rappresenta un fattore di rischio non indipendente per lo sviluppo o la progressione del glaucoma, ma la pachimetria è strettamente correlata anche alla misurazione della pressione oculare: valori pachimetrici che deviano significativamente dai valori medi riscontrati nella popolazione generale possono infatti indurre delle sovrastime o delle sottostime della pressione intraoculare; in particolare, cornee spesse forniranno pressioni più alte del valore reale, cornee sottili forniranno pressioni più basse del valore reale.
Gonioscopia
Rappresenta l’esame diretto dell’angolo irido-corneale, ossia la regione della porzione anteriore dell’occhio (detta camera anteriore) dove l’iride e la cornea si incontrano. Questa regione anatomica è di fondamentale importanza per la fisiologia oculare in quanto è qui che risiedono le strutture deputate al drenaggio dell’umor acqueo, ossia il liquido che circola all’interno dell’occhio e la cui pressione idrostatica determina la pressione intraoculare . Nei glaucomi da chiusura d’angolo, questa regione anatomica risulta essere ridotta in ampiezza, tale da ostacolare il deflusso dell’umor acqueo e il mantenimento della pressione intraoculare fisiologica. L’esame necessita dell’applicazione di una lente apposita sulla superficie dell’occhio, previa instillazione di collirio anestetico, e di osservazione dell’angolo irido-corneale alla lampada a fessura. La gonioscopia dovrebbe essere eseguita almeno una volta in tutti i pazienti con diagnosi di glaucoma o con glaucoma sospetto, in particolare nei pazienti a rischio di glaucoma da chiusura d’angolo.
Secondo le attuali linee guida, l’obiettivo della terapia del glaucoma non è semplicemente la riduzione della pressione oculare: il traguardo che ci si pone è quello di migliorare la qualità di vita del paziente glaucomatoso, o a rischio di glaucoma, a un costo sostenibile per la società. Per questo la decisione su quale terapia utilizzare e su quando iniziarla va personalizzata per ogni paziente. Questo perché non solo la malattia, ma anche la terapia stessa ha un impatto sulla qualità di vita del paziente, nonché sui costi per la società. Attualmente, il medico oculista non ha modo di guarire il danno glaucomatoso quando questo si è già reso manifesto. Può soltanto rallentare la progressione, e questo attualmente si può ottenere solo riducendo la pressione oculare fino a quel valore ideale, caratteristico per ogni paziente (IOP target), raggiunto il quale la riduzione delle cellule ganglionari retiniche diventa simile a quella che si ha fisiologicamente con l’avanzare dell’età. Le armi a disposizione per ridurre la progressione del danno da glaucoma sono la terapia medica, quella laser e quella chirurgica.
La terapia medica prevede l’instillazione quotidiana di farmaci in collirio. Le classi di farmaci utilizzate nella terapia del glaucoma sono gli analoghi delle prostaglandine, i beta bloccanti, gli inibitori dell’anidrasi carbonica, gli alfa-2 agonisti e i parasimpaticomimetici. È stata identificata anche una categoria più recente, gli inibitori delle rho chinasi. In casi selezionati, e generalmente nel breve termine, possono essere utilizzati anche farmaci con modalità di somministrazione diversa da quella in collirio, come il mannitolo, diuretico osmotico somministrato per via endovenosa, o l’acetazolamide in compresse. Ogni classe di farmaci ha uno specifico profilo per quanto concerne gli effetti collaterali e l’efficacia ipotonizzante: si va da una riduzione della pressione oculare da un minimo del 20% a un massimo del 35-40% per singolo principio attivo e possono essere utilizzate, da sole o in combinazione, fino al raggiungimento della IOP target. Affinché la terapia medica sia efficace è fondamentale che il medico oculista responsabilizzi il paziente e gliene spieghi l’importanza, nonché gli effetti collaterali che potrebbe aspettarsi; è altrettanto importante che il paziente sia in grado di autosomministrarsi il collirio, o farselo somministrare da terzi, con precisione e costanza. Tuttavia, se la terapia medica non è ben tollerata o non si dimostra sufficiente ad impedire la progressione della malattia, si pone indicazione alla chirurgia.
La terapia laser prevede la trabeculoplastica laser selettiva (SLT) e la trabeculoplastica argon laser (ALT). Entrambe non necessitano di ospedalizzazione, possono essere eseguite in ambulatorio e permettono una riduzione della pressione oculare media fino al 25%. Possono essere utilizzate come unica terapia, o in associazione a terapia medica o chirurgica. Non sono indicate nelle forme di glaucoma ad angolo chiuso, infiammatorie o neovascolari. Anche l’attacco acuto di glaucoma si giova della terapia laser: se presente un blocco pupillare, questo può essere risolto da un’iridotomia YAG laser, che permette di ristabilire la normale idrodinamica tra la camera posteriore e anteriore dell’occhio. La tecnica chirurgica tradizionale del glaucoma, nonché quella tuttora riconosciuta come gold standard, è la trabeculectomia. L’intervento prevede di creare una fistola tra la camera anteriore dell’occhio e lo spazio sotto la congiuntiva per permettere una via alternativa di deflusso dell’umore acqueo tramite una bozza filtrante. Altra possibilità è l’impianto di una valvola o di un tubo drenante, che solitamente vengono riservati ai casi più complessi, dove la chirurgia tradizionale ha fallito o rischia di fallire per la cicatrizzazione della bozza filtrante.
Altre tecniche possibili sono la sclerectomia profonda, la viscocanalostomia e la canaloplastica. Queste, se da una parte sono meno invasive rispetto alla trabeculectomia, dall’altra sembrerebbero avere una minore efficacia ipotonizzante e sono pertanto meno praticate.
Un’altra possibilità sono le cosiddette MIGS, acronimo per minimally invasive glaucoma surgeries. Queste prevedono l’utilizzo di diversi dispositivi che possono incentivare il deflusso dell’umor acqueo tramite vie diverse, bypassando il trabecolato per permettere all’acqueo di raggiungere il canale di Schlemm, dilatando il canale stesso, oppure oltrepassandolo direttamente per creare uno spazio di deflusso sottocongiuntivale. Queste tecniche prevedono una minore manipolazione chirurgica, un più rapido recupero postoperatorio e un tasso di complicanze piuttosto ridotto. Per contro, sembrano avere un’efficacia ipotonizzante minore rispetto all’intervento classico di trabeculectomia. Vengono di solito riservati a pazienti sui quali non è necessaria una drastica riduzione della pressione oculare. È importante comunque ricordare che la scelta del trattamento va attentamente calibrata per ogni paziente in base al quadro clinico e spetta sempre al chirurgo.
Da ricordare, infine, che anche la chirurgia della cataratta può contribuire enormemente alla riduzione della pressione oculare nelle chiusure angolari indotte dal cristallino.
Da cosa è determinata la pressione degli occhi?
La pressione intraoculare (IOP) dipende principalmente dalla quantità di un liquido prodotto all’interno dell’occhio, chiamato umor acqueo. In pratica, più liquido è presente nel bulbo oculare, maggiore sarà la pressione.
Con quale unità si misura?
Il valore della pressione interna dell’occhio, sebbene differente dalla pressione arteriosa, viene misurato con la stessa unità: i millimetri di mercurio (mmHg). Tradizionalmente, l’oculistica considerava i due valori completamente indipendenti. Tuttavia, studi recenti hanno evidenziato l’esistenza di una certa correlazione: secondo alcuni ricercatori, chi ha una pressione arteriosa elevata potrebbe avere maggiori probabilità di sviluppare una pressione intraoculare alta, anche se la relazione rimane relativamente debole [5].
Qual è il valore massimo tollerabile dall’organismo? 
La pressione intraoculare dovrebbe normalmente essere compresa tra 10 e 20 millimetri di mercurio (mmHg). Il glaucoma è generalmente associato a valori superiori a 20-21 mmHg, ma esiste anche una forma a bassa pressione, in cui il nervo ottico subisce danni pur con valori pressori considerati normali. Questo tipo di glaucoma sembra legato a un ridotto afflusso di sangue al nervo ottico, che provoca una progressiva atrofizzazione delle fibre nervose. Inoltre, nella valutazione della pressione oculare è importante considerare lo spessore della cornea e l’eventuale presenza di una miopia elevata.
Come si misura la pressione intraoculare?
Esistono diversi metodi per misurare la pressione intraoculare, e nel corso degli anni le tecniche si sono evolute, raggiungendo maggiore precisione. Attualmente, negli ospedali, lo strumento più diffuso è il tonometro ad applanazione di Goldmann, che misura la resistenza del bulbo esercitando una leggera pressione sulla cornea. Per gli screening di massa, invece, si utilizza più comunemente il tonometro a soffio, che non richiede contatto diretto con l’occhio, ma sfrutta un getto d’aria indirizzato sulla cornea. (Vedi tonometria).
Quali possono essere altri esami utili?
Può essere utile misurare lo spessore della cornea mediante un esame strumentale chiamato pachimetria, per determinare il valore “reale” della pressione oculare. In particolare, nelle persone con una cornea sottile (pachimetria più bassa) è necessario aumentare di alcuni punti la pressione rilevata per ottenere una misurazione corretta (reale). Al contrario, negli individui con cornea più spessa (pachimetria più alta) bisogna sottrarre alcuni punti dalla rilevazione tonometrica.
Quando bisogna effettuare la pachimetria?
È sufficiente eseguire la pachimetria corneale una sola volta, dal momento che lo spessore della cornea non si modifica in maniera significativa nel tempo, a differenza di altri esami quali il campo visivo o l’OCT che devono essere invece ripetuti periodicamente per monitorare l’eventuale evoluzione del glaucoma.
Come si tratta il glaucoma?
Si cura di solito con colliri per abbassare la pressione oculare (detti “ipotonizzanti”). Per risultare efficace la terapia deve essere eseguita regolarmente e con costanza. In alcuni casi, la terapia può provocare effetti indesiderati: alcune gocce possono causare bruciore, arrossamento degli occhi o mal di testa, generalmente transitori e destinati a scomparire dopo poche settimane. Talvolta possono verificarsi anche piccole alterazioni del ritmo cardiaco, di solito di scarsa rilevanza. Chi dovesse accusare fastidi o disturbi deve sempre informare il proprio medico oculista.
Le persone affette da glaucoma necessitano di controlli periodici, poiché la malattia può peggiorare senza dare sintomi evidenti. In questi casi può essere necessario modificare la terapia. Una volta che il danno al nervo ottico si è verificato, non è più reversibile: si ricorre quindi a farmaci e, se necessario, alla chirurgia (come la trabeculectomia) per cercare di preservare la funzionalità visiva residua.
Il trattamento del glaucoma è efficace solo se la terapia prescritta dal medico oculista viene seguita scrupolosamente. In alcuni casi possono essere consigliati integratori alimentari che contribuiscono alla protezione del nervo ottico. La terapia non deve mai essere sospesa senza consultare l’oculista e il medico di famiglia dovrebbe sempre essere informato dei farmaci in uso. Se i colliri non riescono a controllare adeguatamente la pressione intraoculare, può rendersi necessario ricorrere alla chirurgia o al laser. Le complicanze di questi interventi sono rare, e nella maggior parte dei casi consentono di impedire la progressione della malattia, che altrimenti potrebbe portare a ipovisione o cecità.
Quand’è più probabile che una persona sia affetta da glaucoma?
Quando ha più di quarant’anni e ci sono altri casi in famiglia (per questo si parla di “familiarità” per il glaucoma). Più si è anziani e più aumenta il rischio di essere colpiti da questa patologia oculare detta anche “silente” perché non dà sintomi particolari nelle fasi iniziali. Soprattutto chi ha altri familiari con glaucoma dovrebbe sottoporsi indicativamente a un controllo oculistico una volta l’anno anche in assenza di altre patologie (con misurazione della pressione oculare). La maggiore incidenza di glaucoma sarebbe favorita anche dalla presenza di miopia o ipermetropia elevate, dal diabete e da eventuali terapie protratte a base di cortisonici.
[1] I dati sono stati pubblicati nel 2010. Più recentemente, nel 2017, il Vision Loss Expert Group ha citato la cifra di 60 milioni (AAVV, “Global causes of blindness and distance vision impairment 1990–2020: a systematic review and meta-analysis”, Lancet Glob Health. 2017 Dec;5(12):e1221-e1234. doi: 10.1016/S2214-109X(17)30393-5. Epub 2017 Oct
[2] Fonte: IAPB Internaz.
[3] Fonte: Tham YC, Li X, Wong TY, Quigley HA, Aung T, Cheng CY, “Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis“, Ophthalmology. 2014 Nov;121(11):2081-90. doi: 10.1016/j.ophtha.2014.05.013. Epub 2014 Jun 26. Review.
[4] “Diffuse anomalie strutturali e funzionali nel cervello di un glaucomatoso possono essere, almeno parzialmente, indipendenti da una IOP più elevata (=pressione intraoculare, ndr) e dalla conseguente degenerazione retinica”, cit. tratta dall’articolo di Giorgio A, Zhang J, Costantino F, De Stefano N, Frezzotti P, “Diffuse brain damage in normal tension glaucoma“, Human Brain Mapping, 2017 Oct 24. doi: 10.1002/hbm.23862 (Epub ahead of print)
[5] Horwitz A, Klemp M, Jeppesen J, Tsai JC, Torp-Pedersen C, Kolko M, “Antihypertensive Medication Postpones the Onset of Glaucoma: Evidence From a Nationwide Study“,
Hypertension. 2017 Feb;69(2):202-210. doi: 10.1161/HYPERTENSIONAHA.116.08068. Epub 2016 Dec 5
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus.
Ultima revisione scientifica: 4 marzo 2026.
Leggi le condizioni generali di consultazione di questo sito.











