Lo “scotoma” (dal greco skótos, “oscurità”) è un difetto lacunare del campo visivo. E’ correlato a una riduzione della sensibilità retinica (scotoma relativo) o a una scomparsa completa della sensibilità stessa (scotoma assoluto) in alcune aree della retina, dove l’immagine non risulta più percepibile oppure se ne ha una percezione sbiadita.
Infatti, con uno scotoma assoluto in certe aree del campo visivo la percezione visiva è perduta (si presentano zone scure anche a macchia di leopardo), mentre con uno scotoma relativo in alcune aree non si ha più la percezione cromatica (non si percepiscono più alcuni colori oppure tutti ad eccezione del bianco). Gli scotomi possono avere diverse forme, che variano in base alla patologia di base che li ha determinati. In particolare possono essere: tondeggianti, anulari, ovali, a mosaico.
Come si manifesta?
All’esame del campo visivo (mappa campimetrica) lo scotoma viene rappresentato graficamente come un’area nera che può avere diverse localizzazioni (periferiche o centrali).
Solitamente non viene notato da chi ne è colpito, a meno che ciò non influenzi la visione centrale o non interferisca con l’acuità visiva. Viene definito “negativo” quando non si percepiscono, del tutto o in parte, gli oggetti fissati a causa di una macchia scura. Al contrario è detto “positivo” quando si percepisce una macchia a luminosità intermittente e di colore variabile che si proietta sugli oggetti fissati.
Quali sono le cause?
Le cause sono varie e coinvolgono diverse strutture dell’occhio. Le ragioni principali per cui può presentarsi uno scotoma sono:
alterazioni del nervo ottico (glaucoma, otticopatie, neuriti) e/o lesioni localizzate a livello delle vie ottiche (dalla retina ai centri visivi corticali);
problemi vascolari (spasmi circolatori tipo dell’arteria retinica, emorragie retiniche);
opacità centrale del cristallino (cataratta nucleare).
Quali sono i sintomi?
In linea di massima lo scotoma non comporta disturbi importanti (a meno che non sia di dimensioni notevoli o localizzato nella zona centrale), tanto che in alcune patologie (tipo il glaucoma) ci si rende conto della sua presenza solo quando il campo visivo è già danneggiato e si è, quindi, ristretto. Per scongiurare quest’eventualità è consigliabile sottoporsi periodicamente a un controllo oculistico.
Come si classifica?
Si può classificare in:
scotoma fisiologico (o macchia cieca di Mariotte): zona di non visione che corrisponde al punto in cui emerge il nervo ottico dalla retina (macchia cieca) che, in alcune patologie (tipo il glaucoma e l’edema della papilla), può ampliarsi;
scotoma centrale: coinvolge la zona centrale del campo visivo (entro i 5°) ossia quella che si usa per fissare gli oggetti. Si manifesta quand’è presente un’alterazione funzionale della macula: chi ne è affetto lamenta la visione di una macchia scura al centro. Lo scotoma centrale si può presentare sia a causa di una degenerazione maculare legata all’età (AMD) sia per situazioni infettive tipo toxoplasmosi o focolai di corioretinite oppure, ancora, in caso di alterazioni della conduzione nervosa (otticopatie);
scotoma paracentrale: area di riduzione parziale o totale della sensibilità luminosa che coinvolge l’area paracentrale di fissazione (zona intorno alla macula). Si riscontra nella patologia maculare o del nervo ottico;
scotoma centrocecale: è uno scotoma centrale inizialmente piccolo che lentamente si allarga, coinvolgendo sia il punto di fissazione centrale (macula) sia la macchia cieca. Si può manifestare nelle patologie del nervo ottico e nelle patologie metaboliche da accumulo;
scotoma anulare: area cieca del campo visivo di forma circolare, generalmente localizzato fra 20° e 40° dal punto di fissazione. Si può manifestare in alcune degenerazioni retiniche tipo la retinite pigmentosa;
scotoma arciforme o di Bjerrum: area cieca del campo visivo di forma arcuata o a semiluna suggestivo di una sofferenza del nervo ottico (glaucoma e otticopatie varie); a partenza dalla macchia cieca, presenta una forma di “C” maiuscola nella media periferia del campo visivo;
scotoma scintillante: presenza di una macchiolina scura davanti agli occhi dalla quale si originano piccole strisce scintillanti e colorate (associato molto spesso ad emicrania);
scotomi periferici: sono in genere legati a retinopatie e corioretinopatie.
Come si diagnostica?
L’esame strumentale che permette di effettuare una diagnosi di scotoma è il campo visivo, che dà informazioni sulla visione centrale e sulla visione periferica. Quest’ultima è indispensabile all’uomo ancor più della visione centrale, in quanto gli consente di apprezzare l’esistenza e la morfologia degli oggetti mobili e immobili che lo circondano, consentendogli l’orientamento spaziale. Inoltre è importante eseguire una valutazione neurologica.
Si può curare?
Se le lesioni sono permanenti (ad esempio alle vie ottiche o al tessuto retinico) non esiste una terapia risolutiva; infatti, quando la causa riguarda danni alle cellule nervose (ad esempio in caso di glaucoma od otticopatie) allo stato attuale delle conoscenze scientifiche non si può fare nulla (anche se varie sperimentazioni sono in corso). In ogni caso, si potrebbe recuperare parte della funzionalità retinica residua mediante un efficace processo riabilitativo.
E’ opportuno però fare prevenzione mediante visite oculistiche di screening per capire quando sia necessario instaurare una terapia che tenga sotto controllo la situazione oculare (ad esempio, nel glaucoma l’utilizzo di colliri ipotonizzanti che abbassano la pressione dell’occhio e prevengono danni al nervo ottico).
Quando, invece, la causa riguarda, ad esempio, il cristallino (presenza di una cataratta) è sufficiente un intervento chirurgico per asportarlo e sostituirlo con una lente artificiale intraoculare (IOL).
Per quanto riguarda le patologie vascolari retiniche, tipo le occlusioni vascolari (arteriosa o venosa), è importante identificare la causa precisa per poi instaurare la terapia farmacologica idonea (farmaci anticoagulanti, antiaggreganti e trombolitici) e l’eventuale terapia laser per prevenire le complicanze.
Nelle patologie retiniche come la degenerazione maculare legata all’età (AMD) è importante stabilire la forma di maculopatia (secca o umida) per instaurare eventualmente una terapia opportuna: nelle forma essudativa le iniezioni intravitreali e la terapia fotodinamica, mentre nella forma atrofica ci si limita all’utilizzo di integratori (a base di luteina, zeaxantina, acidi grassi polinsaturi omega-3 e vitamine) che possono migliorare l’apporto nutritivo al centro della retina (trofismo maculare), anche se attualmente non esistono trattamenti farmacologici efficaci.
Nel distacco di retina l’approccio è di tipo chirurgico, volto a riposizionare il tessuto retinico nella sua sede originaria.
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus Leggi le condizioni generali di consultazione di questo sito
Pagina pubblicata il 25 novembre 2010. Ultimo aggiornamento: 5 aprile 2023
Ultima revisione scientifica: 5 aprile 2023
Contatta l’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Il Numero Verde di consultazione oculistica è attivo dal lunedì al venerdì dalle 10 alle 13. Scrivi nel Forum: un medico oculista ti risponderà gratuitamente.
Si tratta di una malattia oculare caratterizzata dal sollevamento della zona centrale della retina a causa dell’accumulo di liquido sieroso.
In particolare, si evidenzia il distacco delimitato di uno strato retinico intermedio (il neuroepitelio) nella regione maculare, a volte associato a distacco sieroso dell’epitelio pigmentato retinico (EPR). Lo stravaso di siero, dai vasi coroideali, passando attraverso lesioni focali dell’EPR (punti di fuga), si accumula nello spazio sottoretinico causandone il distacco. Si tratta di una patologia sporadica, generalmente monolaterale, che può tuttavia colpire entrambi gli occhi in maniera più o meno simmetrica. In genere il sesso maschile tra i 30 e i 50 anni di età (con tendenza ad una personalità di tipo ansioso) è quello più colpito; le donne affette da CSC hanno generalmente un’età media più alta degli uomini. È meno frequente nella razza nera e può presentarsi in forme particolarmente gravi negli asiatici.
Che cause ha?
Sulle cause che determinano la sierosa centrale non si hanno dati certi. Un elemento significativo, che accomuna quasi tutti coloro che ne sono affetti, è che sembrerebbe essere favorita dello stress (si presenta di solito in persone particolarmente attive e competitive). L’induzione o l’aggravamento della malattia sono stati associati a fattori predisponenti quali: ipertensione arteriosa, trapianto di organi, reflusso gastroesofageo, abuso di alcool, terapia steroidea orale e/o inalatoria, gravidanza e patologie sistemiche come il Lupus Eritematoso Sistemico e la Sindrome di Cushing.
L’esordio della malattia è spesso subdolo ed i sintomi sono generalmente monolaterali. I disturbi visivi più frequentemente rilevati sono:
visione appannata (spesso si ha l’impressione di guardare come attraverso una goccia d’acqua);
presenza di uno scotoma centrale relativo;
metamorfopsie (distorsione delle immagini);
visione più scura;
colori sbiaditi;
riduzione dell’acuità visiva.
All’esame del fondo oculare si osserva tipicamente un sollevamento sieroso della retina neurosensoriale localizzato nella regione maculare. Si possono evidenziare uno o più distacchi sierosi dell’epitelio pigmentato, in associazione o anche in assenza del distacco retinico. Col tempo, in sede sottoretinica, si possono notare piccoli precipitati giallastri di materiale proteico o di derivazione dai segmenti esterni dei fotorecettori nell’area del distacco
Come si effettua una diagnosi?
Per identificare la presenza di una corioretinopatia sierosa centrale è indispensabile sottoporsi ad una visita oculistica completa. In prima istanza, lo specialista attraverso l’anamnesi raccoglie più informazioni possibili sul paziente: età, stile di vita, farmaci assunti, presenza di eventuali patologie sistemiche, modalità e tempi d’insorgenza dei disturbi visivi. Alla visita oculistica l’esame del visus può risultare più o meno alterato, il segmento anteriore e la pressione oculare sono di solito nella norma, all’esame del fondo oculare si evidenziano, in sede maculare, i segni tipici della malattia (vedi paragrafo precedente). Per un approfondimento può essere utile sottoporsi ad esami strumentali specifici.
L’OCT (tomografia a coerenza ottica), è una tecnica diagnostica non invasiva che consente di rilevare e monitorare nel tempo, il sollevamento retinico maculare nei pazienti con CSC. Nelle forme croniche, all’esame OCT è possibile rilevare la presenza di cisti intraretiniche e un ispessimento coroideale. In caso di complicanze, si può evidenziare la comparsa di neovasi, facilmente individuabili con l’angio-OCT, una metodica angiografica semplice e non invasiva, che non ha bisogno dell’ iniezione in vena di coloranti.
La fluorangiografia (FAG), è l’esame che da sempre è stato utilizzato per definire le caratteristiche cliniche della CSC. E’ tipico il rilievo di un “leakage” focale o multifocale a livello dell’epitelio pigmentato retinico. Gli eventuali piccoli distacchi dell’epitelio pigmentato risultano iperfluorescenti e chiaramente rilevabili. Nella CSC cronica la fluorangiografia può evidenziare la presenza di un’ epiteliopatia retinica diffusa.
L’ angiografia con verde di indocianina, consente di acquisire informazioni importanti per monitorare il decorso della malattia. Con l’esame è possibile rilevare la diffusione del colorante in aree più o meno estese della coroide sia in corrispondenza delle alterazioni dell’epitelio pigmentato evidenziate dalla fluorangiografia, che in zone dove l’epitelio appare integro. Un’importante iperpermeabilità di alcune zone della coroide caratterizza i pazienti con CSC, sia nei momenti di attività che di inattività della malattia. L’angiografia con verde di indocianina permette anche di individuare eventuali neovascolarizzazioni occulte che possono simulare o complicare una CSC cronica.
L’autofluorescenza è un esame semplice per valutare nel dettaglio il fondo oculare: consente di evidenziare le aree di degenerazione e atrofia dell’epitelio pigmentato (e dei fotorecettori) come conseguenza della prolungata persistenza di liquido sotto la retina. Rappresenta, pertanto, una metodica importante per riconoscere la cronicità della malattia. Da una semplice immagine in autofluorescenza possiamo renderci conto se siamo di fronte a un processo essudativo recente, oppure che dura da diverso tempo (mesi o anni).
No, non vi è un protocollo terapeutico specifico, perché nessun trattamento medico sino ad oggi si è dimostrato sempre valido. Si consiglia generalmente di ridurre lo stress e di condurre uno stile di vita più tranquillo ma queste, chiaramente, sono indicazioni che non sempre i pazienti riescono a seguire. Svariati tipi di farmaci (tranquillanti, antinfiammatori non steroidei, beta bloccanti) sono stati provati senza grande successo, così come inefficace ed anzi in grado di indurre un peggioramento clinico, si è dimostrato essere il cortisone. In qualche caso sembrerebbe avere una certa utilità l’uso di diuretici che riducendo l’accumulo di fluidi sotto la retina, possono accelerare la risoluzione dei sintomi. Attualmente la letteratura medico-scientifica riporta buoni risultati ottenuti con la fotocoagulazione laser, che andrebbe ad agire proprio sul punto di perdita; ma questa eventualità, data la buona prognosi, deve essere valutata dal singolo oculista con molta attenzione e in casi particolari. Il trattamento laser non è indicato, invece, nei casi in cui il punto di fuga sia foveale o nei casi di punti di fuga multipli. Dobbiamo inoltre ricordare che la fotocoagulazione comporta la distruzione di tessuto retinico con eventuali danni a livello visivo (calo del visus, scotoma localizzato, riduzione della sensibilità al contrasto, alterata percezione dei colori). Esiste anche un’altra opzione terapeutica rappresentata dalla terapia fotodinamica indicata anche in quei casi in cui i punti di fuga si presentino iuxtafoveali o sottofoveali e nelle forme croniche; seppur valida, la terapia fotodinamica non è comunque esente da possibili effetti collaterali importanti quali per esempio l’atrofia dell’EPR e la formazione di scotomi localizzati. Più di recente è stato proposto un trattamento con laser giallo micropulsato, che dovrebbe attivare l’epitelio pigmentato favorendo il riassorbimento del liquido sottoretinico. L’efficacia di questo tipo di laser è variabile da un caso all’altro, risulta comunque essere un trattamento indolore, sicuro e potenzialmente ripetibile. Infine, nei casi in cui la CSC si complichi con una neovascolarizzazione coroideale, è indicato il trattamento con iniezioni intravitreali di farmaci anti-VEGF.
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus Leggi le condizioni generali di consultazione di questo sito
Pagina pubblicata il 11 marzo 2008. Ultimo aggiornamento: 13 febbraio 2025
Ultima revisione scientifica: 13 febbraio 2025
Contatta l’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Il Numero Verde di consultazione oculistica è attivo dal lunedì al venerdì dalle 10 alle 13. Scrivi nel Forum: un medico oculista ti risponderà gratuitamente.
La retinopatia del prematuro (ROP) o retinopatia del pretermine, in passato chiamata “fibroplasia retrolentale”, è una malattia vascolare della retina che si manifesta in neonati prematuri e si presenta, in genere, in tutti e due gli occhi, anche se può avere gradi diversi.
Da cosa è causata?
È provocata dalla formazione di nuovi vasi sanguigni (neovascolarizzazioni) nella periferia retinica conseguenti alla prematurità e al basso peso corporeo alla nascita. In particolare l’ossigenoterapia a cui sono sottoposti i prematuri può determinare la comparsa della malattia. Anche eventuali crisi di apnea, infezioni, trasfusioni e persistenza del dotto di Botallo aumentano il rischio di contrarla. Il peso alla nascita è, comunque, sempre il fattore di rischio più importante: si è visto che i nati con peso inferiore a 1250 grammi hanno un rischio elevato di sviluppare una forma medio-grave di retinopatia del prematuro.
Quali sono i sintomi?
Il neonato non è in grado, ovviamente, di riferire alcun sintomo. L’unico segno di malattia può essere la presenza di un riflesso bianco (leucocoria) che, tuttavia, si può osservare solo nelle fasi più avanzate e gravi. Per questo è fondamentale che tutti i nati prematuri siano sottoposti a visite frequenti (anche settimanali) del fondo oculare durante le prime settimane di vita, in modo da poter diagnosticare il prima possibile eventuali segni della patologia; nel caso in cui vengano riscontrati è raccomandabile valutare giorno per giorno l’evoluzione del quadro clinico. L’evoluzione clinica è rapida e può portare a un distacco di retina totale, con conseguente cecità.
Come si effettua la diagnosi?
Attraverso l’esame del fondo oculare. La malattia presenta diversi stadi; purtroppo negli stadi più avanzati si ha un recupero scarso, mentre per le forme iniziali è importantissimo che il controllo sia frequente e venga eseguito da oculisti specializzati. Questo perché anche impercettibili alterazioni delle periferia retinica possono evolvere nel giro di pochi giorni in quadri più gravi.
Che terapie sono disponibili?
Attualmente esistono dei protocolli terapeutici stabiliti dalla comunità scientifica che prevedono determinati trattamenti a seconda dello stadio di malattia. Negli stadi iniziali si esegue un criotrattamento della retina o un suo trattamento laser: “bruciando” la retina periferica si evita che stimoli la formazione dei nuovi vasi dannosi. Quando si manifestano gli ultimi stadi della malattia è necessario intervenire chirurgicamente con vitrectomia o chirurgia del distacco di retina (piombaggio sclerale).
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus Leggi le condizioni generali di consultazione di questo sito
Pagina pubblicata il 3 dicembre 2007. Ultimo aggiornamento: 7 febbraio 2019.
Ultima revisione scientifica: 25 ottobre 2012.
Contatta l’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Il Numero Verde di consultazione oculistica è attivo dal lunedì al venerdì dalle 10 alle 13. Scrivi nel Forum: un medico oculista ti risponderà gratuitamente.
La retinite pigmentosa è una malattia genetica rara che colpisce la retina di entrambi gli occhi, causando un progressivo deterioramento della funzione visiva. Fa parte di un insieme di patologie ereditarie caratterizzate dalla degenerazione graduale delle cellule retiniche responsabili della percezione della luce. I primi sintomi riguardano generalmente la difficoltà a vedere in condizioni di scarsa illuminazione e la riduzione della visione periferica. Con il passare del tempo, la malattia può interessare anche la visione centrale, compromettendo in modo significativo la capacità visiva complessiva.Conosciuta anche come retinosi pigmentaria o retinopatia pigmentosa, questa patologia determina un progressivo restringimento del campo visivo e una diminuzione dell’acuità visiva. Negli stadi più avanzati può portare a ipovisionesevera e, nei casi più gravi, alla cecità.
Come funziona la retina?
La retina è un sottile strato di tessuto nervoso organizzato in più livelli, rappresenta la componente fondamentale dell’occhio, perché svolge un ruolo centrale nel meccanismo della visione. La luce proveniente dall’ambiente esterno viene focalizzata su questa struttura, dove viene convertita in impulsi elettrochimici. Tali segnali vengono poi trasmessi al cervello attraverso il nervo ottico e qui elaborati fino ad essere convertiti in immagini. Quando le cellule retiniche subiscono un danno, questa catena di trasmissione si interrompe e la capacità visiva risulta compromessa. All’interno della retina sono presenti circa 131 milioni di cellule fotosensibili, chiamate fotorecettori, che si suddividono in due categorie principali:
I coni, così denominati per la loro forma, sono responsabili della percezione dei dettagli e dei colori. Ne esistono tre varietà, ciascuna sensibile a una specifica lunghezza d’onda corrispondente al rosso, al verde e al blu; la combinazione delle loro risposte viene interpretata dal cervello come una determinata tonalità cromatica. I coni sono circa 6 milioni e si concentrano soprattutto nella zona centrale della retina, chiamata macula. Per questo motivo risultano indispensabili per la visione centrale, necessaria ad attività come la lettura, il riconoscimento dei volti e la guida. La loro capacità di discriminare i particolari è enormemente superiore a quella dei bastoncelli.
I bastoncelli, invece, hanno una forma più allungata e rispondono principalmente alle variazioni di luminosità e al movimento. Sono circa 125 milioni e si distribuiscono prevalentemente nelle aree periferiche della retina, dove superano di gran lunga il numero dei coni. Nella regione centrale sono poco presenti, mentre diventano molto numerosi verso la periferia. Grazie ai bastoncelli siamo in grado di percepire oggetti in movimento ai margini del campo visivo, anche senza distinguerne con precisione i dettagli.
Quante persone colpisce la retinite pigmentosa?
La retinite pigmentosa è una malattia rara che colpisce circa una persona ogni 4.000 negli Stati Uniti ed è ancora oggi una delle principali cause di riduzione grave della vista e di cecità nelle persone sotto i 60 anni. In Italia, secondo i dati disponibili, si stima che ne siano affette circa 30.000 persone, mentre in Europa il numero sale a circa 167.000. Se si considerano nel loro insieme tutte le degenerazioni ereditarie della retina, il totale dei soggetti coinvolti a livello mondiale raggiungerebbe i 2 milioni di cui circa 1,5 milioni soffrirebbero proprio di retinite pigmentosa.Attualmente non esiste ancora una cura in grado di risolvere definitivamente la malattia. Tuttavia, numerosi gruppi di ricerca stanno lavorando su possibili terapie, con l’obiettivo di rallentarne l’evoluzione o migliorare la qualità della vita delle persone colpite. La retinite pigmentosa può manifestarsi in età molto diverse: nella maggior parte dei casi compare tra l’adolescenza e l’età adulta, ma non mancano situazioni in cui i primi sintomi si presentano già durante l’infanzia. L’andamento della malattia è generalmente progressivo: la capacità visiva tende a diminuire nel tempo, con un peggioramento graduale che può diventare significativo negli anni. L’unico aspetto sul quale la comunità scientifica concorda pienamente riguarda l’origine genetica della retinite pigmentosa. Si tratta infatti di una patologia ereditaria, trasmessa all’interno delle famiglie secondo modalità ormai ben note. Le ricerche condotte finora hanno permesso di individuare circa un centinaio di geni che, se alterati, possono essere responsabili dell’insorgenza della malattia, spiegando così la grande varietà di forme e gravità con cui essa si presenta.
Quali forme esistono di retinite pigmentosa?
La retinite pigmentosa è una malattia di origine genetica che può essere trasmessa secondo diverse modalità ereditarie. Le forme di trasmissione più comuni sono quella autosomica dominante, autosomica recessiva e quella legata al cromosoma X; in una parte dei casi la patologia si manifesta invece in modo apparentemente sporadico, senza altri casi noti all’interno della stessa famiglia.
Nella trasmissione autosomica dominante, è sufficiente che uno dei due genitori sia affetto dalla malattia perché esista una probabilità del 50% che ciascun figlio erediti la mutazione responsabile. Questa forma colpisce maschi e femmine con la stessa frequenza.
La forma autosomica recessiva si verifica quando entrambi i genitori sono portatori sani della mutazione genetica, pur non presentando sintomi. In questo caso, ogni figlio ha una probabilità del 25% di sviluppare la malattia. Anche questa modalità interessa in egual misura entrambi i sessi.
Esiste poi la trasmissione legata al cromosoma X, nella quale il gene responsabile è localizzato sul cromosoma sessuale X. In questa situazione, la madre è generalmente portatrice sana e può trasmettere la mutazione ai figli maschi, che risultano affetti, con una probabilità del 50%. Le figlie femmine, invece, nella maggior parte dei casi non sviluppano la malattia, ma hanno a loro volta il 50% di probabilità di essere portatrici sane. Di conseguenza, questa forma di retinite pigmentosa colpisce quasi esclusivamente individui di sesso maschile.
In circa il 30% dei casi la retinite pigmentosa viene definita sporadica, poiché si osserva un solo soggetto affetto all’interno della famiglia, anche considerando più generazioni. Tuttavia, questa definizione si basa esclusivamente sull’anamnesi familiare: non sempre è possibile escludere con certezza una trasmissione autosomica recessiva o legata al sesso, soprattutto quando la persona colpita è un maschio.
Infine, quando la retinite pigmentosa si presenta in associazione a una perdita dell’udito, si parla di sindrome di Usher, una condizione genetica complessa che coinvolge sia il sistema visivo sia quello uditivo.
Quali sono i sintomi?
I primi segnali che possono far sospettare la presenza di una retinite pigmentosa sono principalmente legati a difficoltà visive che compaiono in modo graduale. Uno dei sintomi più caratteristici è la ridotta capacità di vedere in condizioni di scarsa illuminazione, soprattutto di sera o di notte. Le persone colpite possono avere problemi a muoversi in ambienti poco illuminati o a guidare dopo il tramonto, e spesso riferiscono un adattamento lento quando si passa da spazi luminosi a luoghi più bui. Questo disturbo è dovuto al fatto che, nelle fasi iniziali della malattia, vengono colpiti soprattutto i bastoncelli, le cellule della retina responsabili della visione notturna.Con il progredire della patologia, si manifesta anche una progressiva riduzione del campo visivo. Diventa sempre più difficile percepire ciò che si trova ai lati, tanto che oggetti laterali, gradini o ostacoli bassi possono non essere notati. Questa condizione, spesso definita “visione tubulare”, tende ad accentuarsi nel tempo e, nei casi più avanzati, può estendersi fino a coinvolgere la parte centrale della retina, compromettendo anche la visione centrale. La velocità con cui si verifica questo peggioramento e l’età di comparsa dei sintomi variano da persona a persona e dipendono da diversi fattori, tra cui il tipo di alterazione genetica alla base della malattia. A questi disturbi si associano frequentemente una maggiore sensibilità alla luce, una riduzione della sensibilità al contrasto e una crescente difficoltà nel riconoscere con chiarezza l’ambiente circostante.
Come si effettua la diagnosi?
La diagnosi di retinite pigmentosa, quando sono presenti i sintomi tipici della malattia, è generalmente agevole ed è di competenza dell’oculista, spesso in collaborazione con il genetista. Il percorso diagnostico si basa su una serie di esami clinici e strumentali mirati a valutare la funzionalità e la struttura della retina, in particolare:
l’esame del visus;
l’osservazione del fondo oculare;
lo studio del campo visivo;
l’elettroretinogramma;
la fluorangiografia retinica.
La malattia può essere identificata in diverse fasi della vita: dall’infanzia all’adolescenza, fino all’età adulta, anche se l’età di esordio varia notevolmente da persona a persona. Nei casi in cui il quadro clinico non sia immediatamente chiaro, la diagnosi si fonda su una valutazione approfondita di tutti gli elementi disponibili, come l’età di comparsa dei sintomi, la modalità di progressione della malattia e l’eventuale presenza di altri disturbi oculari o sistemici. In queste situazioni possono risultare particolarmente utili esami più specifici, come lo studio elettrofisiologico, che comprende l’elettroretinogramma (ERG) e l’elettrooculogramma (EOG), oltre ai test di adattamento al buio. Anche la valutazione della percezione dei colori e la fluorangiografia della retina possono fornire informazioni complementari. Un aspetto importante del processo diagnostico è inoltre l’analisi dell’intero nucleo familiare, necessaria per individuare il tipo di trasmissione ereditaria della malattia e definire correttamente il quadro genetico.
L’esame del visus, consente di valutare l’acuità visiva a livello della parte centrale della retina. Viene eseguito chiedendo al paziente di leggere, su un apposito tabellone posizionato di solito ad una distanza di 3 o 5 metri, caratteri di dimensioni progressivamente più piccole.
L’esame del fondo oculare ha lo scopo di analizzare la struttura della retina e di individuare l’eventuale presenza di depositi pigmentari sulla sua superficie. Nella retinite pigmentosa, tali alterazioni assumono spesso un aspetto caratteristico, comunemente descritto come “a spicole ossee”. È importante sottolineare, tuttavia, che alcune forme di retinite possono manifestare sintomi del tutto simili pur non presentando queste tipiche modificazioni del fondo oculare.
L’esame del campo visivo permette di valutare la sensibilità della retina agli stimoli luminosi nelle diverse aree della retina. Fornisce una documentazione oggettiva delle difficoltà riferite dal paziente, in particolare durante il movimento, e rappresenta uno strumento essenziale per seguire nel tempo l’evoluzione della malattia.
L’elettroretinogramma (ERG) è un esame che consente di registrare l’attività elettrica della retina in risposta a specifici stimoli luminosi. Attraverso questo test è possibile valutare in modo separato la funzionalità dei due principali tipi di fotorecettori, i coni e i bastoncelli. L’ERG riveste un ruolo fondamentale nella diagnosi della retinite pigmentosa, poiché già nelle fasi iniziali della malattia il tracciato risulta spesso marcatamente ridotto o quasi assente, indicando una compromissione precoce della funzione retinica.
La fluorangiografia (FAG) è un esame diagnostico che prevede l’iniezione per via endovenosa di un colorante (fluoresceina), seguita dall’esecuzione di una serie di fotografie della retina in momenti successivi. Attraverso la circolazione sanguigna, il colorante raggiunge i vasi retinici, rendendo visibili arterie, capillari e vene. Questo permette di valutarne la distribuzione e di analizzare lo stato delle pareti vascolari, fornendo informazioni utili sulle condizioni della circolazione retinica.
Che decorso ha la malattia?
Il decorso della retinite pigmentosa è estremamente variabile da persona a persona, ma la malattia ha comunque un andamento progressivo e invalidante. Nella maggior parte dei casi, i disturbi visivi tendono ad accentuarsi nel tempo e il campo visivo si restringe gradualmente, fino a ridursi in alcuni casi in modo marcato. Con l’avanzare della patologia possono comparire anche altri sintomi, come una maggiore sensibilità alla luce, difficoltà nella percezione dei colori e lo sviluppo di cataratta. Purtroppo, nelle fasi più avanzate, l’evoluzione della malattia può condurre, in molti casi, a una grave compromissione della vista fino alla cecità.
Si può curare?
Attualmente la retinite pigmentosa non è considerata una malattia guaribile. In passato, molte speranze erano state riposte nella terapia iperbarica, nota anche come ossigenoterapiaiperbarica(OTI), con l’obiettivo di rallentare o arrestare la progressione della patologia. Diversi studi hanno effettivamente evidenziato una risposta positiva a livello cellulare, confermata anche da esami strumentali come l’elettroretinogramma, che nei pazienti sottoposti al trattamento mostrava un aumento significativo dell’ampiezza del tracciato. Tuttavia, nonostante questi risultati incoraggianti, l’ossigenoterapia non si è dimostrata risolutiva e non interviene sulle cause profonde della malattia.Nonostante i progressi clinici concreti siano stati finora limitati, la ricerca scientifica è molto attiva e segue diversi filoni di studio. Attualmente le prospettive più promettenti riguardano la terapia genica e l’impiego di cellule staminali.
Le vitamine possono aiutare?
Negli ultimi anni è stata osservata una certa efficacia di terapie di supporto basate sull’assunzione di specifici integratori, in particolare nelle fasi iniziali della malattia, quando la visione centrale risulta ancora relativamente conservata. In questo contesto, alcuni studi hanno evidenziato un’associazione favorevole tra l’impiego della vitamina A e una gestione più efficace della retinite pigmentosa, soprattutto se inserita all’interno di un percorso di monitoraggio clinico. Benefici potenziali sono stati osservati anche con l’assunzione di acidi grassi omega‑3, antiossidanti e luteina, sostanze utili a sostenere la salute della retina e a contrastare i meccanismi di stress ossidativo. È fondamentale sottolineare che questi integratori devono essere assunti a dosaggi controllati e sempre sotto supervisione medica, poiché non rappresentano una cura, ma un possibile supporto volto a preservare più a lungo la funzione visiva residua.
Terapia genica
L’obiettivo della ricerca è individuare i geni responsabili della malattia per poter intervenire in modo mirato attraverso le moderne tecniche di ingegneria genetica. In questo ambito si punta, da un lato, a sostituire i geni alterati con copie sane e, dall’altro, soprattutto nelle forme a trasmissione autosomica dominante, a disattivare i geni che risultano dannosi. Per trasportare il materiale genetico corretto viene generalmente utilizzato un virus vettore, opportunamente modificato per renderlo innocuo. Questo virus, paragonabile a un “cavallo di Troia”, è in grado di introdurre all’interno delle cellule le sequenze di DNA necessarie alla correzione del difetto genetico. Attraverso iniezioni effettuate sotto la retina si tenta così di intervenire direttamente sulla causa della malattia. Questo approccio ha già mostrato risultati positivi soprattutto nei bambini affetti da un’altra patologia genetica retinica, l’amaurosi congenita di Leber. Nel caso della retinite pigmentosa, tuttavia, la presenza di un numero molto elevato di geni coinvolti rende il trattamento genetico più complesso e ancora oggetto di studio.Negli ultimi anni la ricerca scientifica ha compiuto importanti passi avanti nella comprensione delle cause genetiche della retinite pigmentosa e nello sviluppo di nuove terapie, in particolare nell’ambito della terapia genica. Nonostante questi progressi, al momento non esiste ancora un trattamento efficace valido per tutti i pazienti. Attualmente l’unica terapia genica approvata è quella con voretigeneneparvovec, indicata però esclusivamente per le forme di retinite pigmentosa causate da una mutazione biallelica del gene RPE65. Parallelamente, la ricerca sta avanzando rapidamente anche per altre varianti della malattia: sono infatti in corso studi clinici in fase avanzata per alcune forme specifiche, come quella legata al cromosoma X associata al gene RPGR, oltre allo sviluppo di tecniche innovative di editing genetico, tra cui CRISPR-Cas9, che aprono nuove prospettive per il futuro.
Cellule staminali
La ricerca sulle cellule staminali applicata alla retinite pigmentosa si trova attualmente in una fase di rapido sviluppo. Sono in corso studi clinici di fase iniziale, mirati principalmente a valutare la sicurezza e l’efficacia di questi approcci terapeutici. Le cellule staminali vengono studiate con due obiettivi principali: da un lato, sostituire i fotorecettori che sono andati perduti a causa della degenerazione della retina; dall’altro, fornire un sostegno alle cellule retiniche ancora vitali, rallentando il processo degenerativo attraverso un effetto protettivo.In particolare, si stanno organizzando studi che utilizzano cellule staminali progenitrici della retina, somministrate mediante iniezione nella cavità vitreale, soprattutto nei casi di retinite pigmentosa in fase avanzata. Parallelamente, l’attenzione dei ricercatori si sta concentrando anche su altre tipologie cellulari, come le cellule staminali neurali e le cellule staminali mesenchimali derivate dalla gelatina di Wharton del cordone ombelicale. Queste ultime si sono dimostrate capaci di rilasciare sostanze protettive in grado di ridurre l’infiammazione e limitare la morte delle cellule retiniche.Un altro ambito di grande interesse è rappresentato dalle cellule staminali pluripotenti indotte, ottenute a partire da cellule adulte. Queste cellule hanno mostrato, nei modelli sperimentali, la capacità di differenziarsi in cellule retiniche funzionali senza evidenziare un rischio significativo di formazione tumorale, aprendo prospettive potenzialmente importanti per il futuro.Nonostante i risultati preliminari siano incoraggianti, la terapia con cellule staminali non è ancora considerata un trattamento standard per la retinite pigmentosa. Restano infatti aperte diverse sfide, in particolare quelle legate alla sicurezza nel lungo periodo e alla reale capacità delle nuove cellule di integrarsi in modo efficace e funzionale con il tessuto retinico esistente.
Trapianto di retina
Attualmente non esiste ancora un vero e proprio trapianto biologico della retina. Tuttavia, la ricerca sta esplorando approcci innovativi che potrebbero aprire nuove prospettive terapeutiche. Tra questi vi sono i sistemi di retina artificiale, basati su microchip in grado di sostituire parzialmente la funzione dei fotorecettori danneggiati. Questi dispositivi sono già utilizzati da alcuni anni e le sperimentazioni hanno dimostrato la capacità di riconoscere oggetti o contorni, migliorando in parte l’autonomia dei pazienti, anche se i risultati ottenuti non sono sempre pienamente soddisfacenti.Un’altra strategia emergente è rappresentata dalla cosiddetta “retina liquida”, un prototipo già testato con risultati promettenti, che prevede l’iniezione di nanoparticelle capaci di comportarsi in modo simile ai fotorecettori naturali. Anche questo approccio è ancora in fase sperimentale, ma offre interessanti prospettive per il futuro trattamento delle forme più avanzate della malattia.
Immunologia
Si prefigge di verificare alcune teorie che ipotizzerebbero un’alterazione del sistema immunitario che potrebbe essere il principale fattore scatenante della malattia. Le ricerche più recenti in ambito immunologico hanno messo in evidenza il ruolo fondamentale dell’infiammazione nella progressione della retinite pigmentosa, aprendo nuove prospettive terapeutiche. Diversi studi indicano infatti che, nella retina in fase degenerativa, si attivano processi infiammatori che contribuiscono alla morte dei fotorecettori, indipendentemente dalla specifica mutazione genetica responsabile della malattia. La perdita dei fotorecettori innesca una risposta immunitaria locale che coinvolge le cellule gliali, normalmente deputate al supporto delle cellule nervose. Tuttavia, quando questa attivazione diventa cronica, le cellule gliali possono rilasciare sostanze potenzialmente dannose, contribuendo ad accelerare il processo degenerativo. In alcuni casi, il sistema immunitario può arrivare ad aggredire erroneamente le cellule retiniche già compromesse, aggravando ulteriormente il danno.Alla luce di queste evidenze, la ricerca attuale si sta concentrando sul possibile riutilizzo di farmaci già disponibili per contrastare la componente infiammatoria della malattia. In particolare, studi recenti suggeriscono che l’impiego di farmaci antinfiammatori come i glucocorticoidi, potrebbero rallentare la perdita della funzione visiva e favorire la sopravvivenza dei coni, indipendentemente dal gene coinvolto. L’obiettivo di questo approccio non è correggere direttamente la causa genetica della retinite pigmentosa, ma proteggere i fotorecettori ancora integri, contribuendo a rallentare la progressione della malattia e a preservare più a lungo la funzione visiva residua.
Il futuro dei trattamenti
Come già accennato in precedenza, le possibilità di cura della retinite pigmentosa sembrano essere sempre più legate allo sviluppo della terapia genica e all’impiego delle cellule staminali. In alcuni pazienti che hanno perso la vista a causa della malattia sono stati impiantati microchip nella retina. Quando l’intervento ha avuto esito positivo, questi dispositivi hanno consentito di recuperare una forma di visione molto limitata, ma sufficiente in alcuni casi a migliorare l’autonomia e l’orientamento nello spazio.
Accanto alle nuove tecnologie terapeutiche, ancora in fase sperimentale, sono oggi disponibili numerose soluzioni di tipo assistivo pensate per le persone ipovedenti, con l’obiettivo di migliorare la qualità della vita quotidiana. Molte di queste tecnologie sono integrate in dispositivi di uso comune, come computer, smartphone, tablet che offrono funzioni quali l’ingrandimento delle immagini, l’aumento del contrasto tra chiaro e scuro e la conversione dei testi in formato audio. Alcune applicazioni consentono inoltre di utilizzare la fotocamera dello smartphone per riconoscere e descrivere gli oggetti inquadrati, facilitando l’orientamento nello spazio e la mobilità. Quando la funzione visiva non è completamente compromessa, possono essere intrapresi specifici percorsi di riabilitazione visiva. Questi trattamenti non mirano a ripristinare la vista, ma a gestire in modo più efficace la progressione della malattia e a valorizzare al massimo la capacità visiva residua, sfruttando i fotorecettori ancora funzionanti.
Nelle forme più avanzate di retinite pigmentosa, assume inoltre un’importanza fondamentale il supporto psicologico. Affrontare una diagnosi di questo tipo, insieme alla progressiva perdita della funzione visiva, può avere un forte impatto emotivo e comportare un aumento significativo del rischio di sviluppare disturbi dell’umore, come stati depressivi. Per questo motivo, è spesso consigliato intraprendere un percorso di sostegno psicologico, che può includere colloqui individuali o il coinvolgimento della famiglia, con l’obiettivo di favorire l’adattamento alla malattia, ridurre il senso di isolamento e migliorare il benessere complessivo della persona.
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus Leggi le condizioni generali di consultazione di questo sito
Ultima revisione scientifica: 14 maggio 2026
Contatta l’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Il Numero Verde di consultazione oculistica è attivo dal lunedì al venerdì dalle 10 alle 13. Scrivi nel Forum: un medico oculista ti risponderà gratuitamente.
Si tratta di una malattia degenerativa oculare che consiste in una crescita anomala di tessuto fibroso e vascolare della congiuntiva bulbare, che arriva a coprire la superficie esterna e trasparente dell’occhio che si trova davanti all’iride (cornea). Si potrebbe considerare una sorta di “callo” oculare morbido. Lo pterigio si sviluppa piuttosto lentamente nel corso degli anni e la sua evoluzione non è costante, bensì caratterizzata da periodi di stabilità e periodi in cui la crescita e più rapida. Quando lo pterigio arriva ad invadere la cornea, quest’ultima appare biancastra e ricca di vasi, con una superficie non regolare. Tale situazione induce spesso una serie di fastidiosi sintomi per il paziente, oltre che una riduzione significativa del visus.
Qual è la causa?
Diversi studi hanno messo in evidenza il ruolo svolto dai fattori ambientali nella patogenesi dello pterigio, in particolare hanno evidenziato una correlazione con l’esposizione quotidiana e prolungata ai raggi ultravioletti (UV). Per tale motivo i soggetti maggiormente esposti al rischio di sviluppare uno pterigio sono i marinai, gli agricoltori, gli alpinisti, gli edili, quindi tutte quelle persone che trascorrono diverse ore al giorno all’aria aperta, esponendosi ai raggi del sole, senza utilizzare appositi sistemi di protezione (occhiali scuri contro i raggi UV, cappellini con visiera). Ci sono anche altri fattori di rischio che sembrerebbero essere coinvolti nell’origine dello pterigio, quali: alterazioni importanti del film lacrimale, stati irritatitvi cronici della superficie oculare, razza, ereditarietà. Infine, va ricordato che lo pterigio prevale nei soggetti di sesso maschile e tende a manifestarsi più frequentemente nei soggetti che hanno raggiunto i 40 anni d’età.
Quali sono i sintomi?
Lo pterigio può essere privo di sintomi specifici, soprattutto nelle fasi iniziali, in caso di situazioni più gravi e/o in presenza d’infiammazione, si presentano frequentemente: arrossamento, bruciore, lacrimazione eccessiva, senso di abbagliamento durante la guida notturna o in occasione di esposizione a fonti di luce artificiale, sensazione di avere un corpo estraneo nell’occhio. Quando lo pterigio si accresce si evidenzia una riduzione del visus che fondamentalmente dipende da due fattori: insorgenza di un astigmatismo, dovuto all’estensione dello pterigio sulla cornea con conseguente deformazione della sua curvatura ; invasione della zona ottica (viene coperta la pupilla).
Quali sono i segni?
È possibile vedere anche ad occhio nudo, oltre che con la lampada a fessura, la presenza del tessuto congiuntivale anomalo sulla superficie oculare. Si presenta come un triangolo (in cui si possono distinguere la testa, il collo e il corpo), con l’apice (testa) rivolto verso il centro della cornea. Con la lampada a fessura si può constatare l’abbondanza di vasi. In condizioni d’infiammazione il diametro vascolare è maggiore.
Qual è la terapia?
La terapia è chirurgica: non esistono altre modalità di cura per lo pterigio. Si tratta di un intervento semplice, che si effettua in anestesia locale (con chirurgia ambulatoriale). È indicato soprattutto nei seguenti casi: astigmatismi non correggibili, occlusione della zona ottica, infiammazioni ricorrenti non controllabili con la terapia locale e, in ultimo, per motivi estetici. L’intervento spesso recidiva: lo pterigio può riformarsi. Fondamentale è la prevenzione dello pterigio per chi si espone ai raggi ultravioletti. L’utilizzo di occhiali da sole a norma di legge, infatti, oltre a prevenirne la comparsa, protegge anche le strutture oculari dai potenziali danni delle radiazioni UV (in particolare la retina e il cristallino).
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus Leggi le condizioni generali di consultazione di questo sito
Pagina pubblicata il 1 luglio 2008. Ultimo aggiornamento: 24/01/2023
Ultima revisione scientifica: 24 gennaio 2023
Contatta l’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Il Numero Verde di consultazione oculistica è attivo dal lunedì al venerdì dalle 10 alle 13. Scrivi nel Forum: un medico oculista ti risponderà gratuitamente.
L’oncocerchiasi (oncocercosi), detta anche “cecità dei fiumi”, è causata dall’infezione provocata da un verme (nematode), l’Onchocerca volvulus che si riproduce vicino i maggiori corsi d’acqua che attraversano molti paesi dell’Africa equatoriale e dell’America meridionale. Le larve dell’agente vettore della malattia, si sviluppano solamente in acque limpide, molto ossigenate e con scarse sostanze organiche. Il termine “cecità dei fiumi” indica la relazione tra la malattia a livello oculare e l’area abitata dai soggetti malati. Le piccole larve si trasformano in vermi adulti e possono vivere anche 15 anni; questo lungo periodo di vita ne facilita la trasmissione tra gli esseri umani. Ogni giorno i vermi femmina producono migliaia di larve microscopiche, dette microfilarie, che si diffondono nel corpo attraverso la puntura delle mosche sull’uomo e quando muoiono causano forti reazioni come infiammazione, irritazione e prurito. Se le larve raggiungono gli occhi possono verificarsi danni irreversibili alla vista. In particolare, viene infettata la cornea (si sviluppa una cheratite sclerosante) che spesso si accompagna a un’infiammazione interna dell’occhio con conseguenti danni alla retina dovuti a cicatrizzazione e degenerazione del tessuto nervoso (atrofia). Si ritiene che l’oncocercosi sia, dopo il tracoma, la seconda causa al mondo di cecità prevenibile tra le malattie contagiose. Tuttavia va detto che, grazie agli sforzi mondiali, la patologia sembra essere in via di progressiva riduzione.
Epidemiologia
L’oncocercosi colpisce principalmente le popolazioni rurali dell’Africa subsahariana e dello Yemen, con focolai endemici più piccoli in alcune parti dell’America Latina. Si manifesta di più negli uomini che nelle donne e ha un’incidenza minore nella fascia d’età 0-10 anni, con un picco maggiore in soggetti di 20-40 anni. Sono infatti soprattutto le persone sopra i 40 anni a poter essere affette dalla cecità irreversibile. L’infestazione dei vermi avviene durante l’infanzia, senza che i soggetti manifestino segni di malattia per lunghi periodi di tempo inoltre, per essere infettati bisogna essere punti diverse volte e soggiornare nelle zone ad alto rischio per più di tre mesi. Il numero di vermi adulti presenti in un soggetto è relativo al numero di punture fatte dalla mosca nera, pertanto i rischi d’ infezione sono direttamente proporzionali al numero di punture ricevute nella vita.
Diagnosi
La diagnosi di oncocercosi può essere eseguita in qualsiasi fase della malattia, ma bisogna intervenire in tempo per scongiurare danni irreparabili. In particolare la diagnosi si può fare in due modi:
con la conta delle larve (microfilarie);
visualizzando la larva adulta (dopo aver eseguito la biopsia ad un nodulo sottocutaneo oppure individuando le microfilarie all’interno dell’occhio).
Clinica
Il primo segno della malattia è una modesta uveite anteriore (infiammazione oculare). Il piccolo parassita biancastro (“vermetto”), che si trova nella parte dell’occhio più vicina alla pupilla (detta “camera anteriore”), può essere individuato all’esame con lampada a fessura. Con l’andare del tempo e con il persistente ingresso delle microfilarie all’interno dell’occhio, l’infiammazione progredisce e la situazione si aggrava a causa dei fenomeni di cicatrizzazione.
Il coinvolgimento del segmento posteriore dell’occhio produce una corioretinite (infiammazione della coroide e della retina). L’ulteriore progressione della patologia porta alla comparsa di atrofia corioretinica (morte e perdita di tessuto retinico e cicatrizzazione), accumulo di pigmento, fibrosi e neovascolarizzazione (proliferazione di nuovi vasi sanguigni). Le lesioni sono generalmente simmetriche (coinvolgono entrambi gli occhi) e tendono a risparmiare il centro della retina (la macula) fino agli stadi terminali della malattia.
Terapia
L’oncocercosi può essere trattata con una dose annuale di ivermectina (un antiparassitario) per almeno 10-15 anni; il farmaco si assume per via orale, di solito sotto forma di un’unica compressa da deglutire. È stato anche sperimentato un altro antiparassitario, il moxidecin, da assumere in dose singola di 8 mg per via orale. Ovviamente oltre al trattamento farmacologico, il modo ideale per trattare la patologia è quello di prevenirla.
Programmi OMS di cura e prevenzione
Oltre il 99% delle persone infette vive in Africa e nello Yemen; il restante 1% vive al confine tra Brasile e Venezuela. Nel 2023 almeno 249,5 milioni di persone hanno richiesto un trattamento preventivo contro l’oncocercosi. Il Global Burden of Disease Study ha stimato nel 2017 che 14,6 milioni di persone infette avevano già una malattia della pelle e 1,15 milioni avevano perso la vista.
L’OMS ha verificato che cinque paesi sono indenni dall’oncocercosi dopo aver implementato con successo attività di eliminazione per decenni: quattro nella regione delle Americhe: Colombia (2013), Ecuador (2014), Messico (2015) e Guatemala (2016), e uno in Africa: Niger (2025).
Tra il 1974 e il 2002, l’oncocercosi è stata tenuta sotto controllo nell’Africa occidentale attraverso il lavoro dell’Onchocerciasis Control Programme (OCP), utilizzando principalmente l’irrorazione di insetticidi contro le larve di mosca nera (controllo dei vettori) da parte di elicotteri e aerei. Questo è stato poi integrato dalla distribuzione su larga scala di ivermectina dal 1989. Il programma OCP ha guarito 40 milioni di persone dall’infezione, ha prevenuto la cecità in 600 mila persone e ha assicurato a 18 milioni di bambini una nascita senza questa malattia.
L’African Programme for Onchocerciasis Control (APOC) è stato lanciato nel 1995 con l’obiettivo di controllare l’oncocercosi nei restanti paesi endemici in Africa e si è chiuso alla fine del 2015 dopo aver iniziato la transizione verso l’eliminazione dell’oncocercosi. La sua strategia principale era l’istituzione di un trattamento sostenibile diretto alla comunità con ivermectina e il controllo dei vettori con metodi sicuri per l’ambiente, ove appropriato [1].
Sono stati istituiti comitati nazionali per l’eliminazione dell’oncocercosi (NOEC) in 25 paesi in Africa per sviluppare e implementare nuove strategie.
Il Global Onchocerciasis Network for Elimination (GONE) è stato lanciato a gennaio 2023 dall’OMS, dai suoi stati membri e dai partner, il cui obiettivo è supportare i paesi nell’accelerare i progressi verso il raggiungimento degli obiettivi della road map per l’eliminazione dell’oncocercosi.
L’OMS in azione
La Roadmap dell’OMS per le malattie tropicali neglette (NTD) 2021-2030 ha identificato l’oncocercosi come una delle malattie da eliminare. La Roadmap ha fissato obiettivi ambiziosi da raggiungere entro il 2030, ovvero eliminare la necessità di MDA (somministrazione di massa di farmaci) di ivermectina in almeno 1 focus in 34 paesi, in oltre il 50% della popolazione in almeno 16 paesi e nell’intera popolazione endemica in almeno 12 paesi. L’Onchocerciasis Technical Advisory Subgroup (OTS) istituito dall’OMS nel 2017 fornisce indicazioni e supervisione per la ricerca operativa per identificare le aree endemiche che richiedono MDA [1].
Pagina pubblicata il 30 giugno 2009. Ultimo aggiornamento: 3 settembre 2025.
Ultima revisione scientifica: 25 marzo 2025.
Contatta l’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Il Numero Verde di consultazione oculistica è attivo dal lunedì al venerdì dalle 10 alle 13. Scrivi nel Forum: un medico oculista ti risponderà gratuitamente.
La neuropatia ottica ischemica anteriore (NOIA) è una patologia acuta e indolore che può causare una perdita visiva improvvisa e spesso permanente. La condizione si verifica quando il flusso sanguigno al nervo ottico viene interrotto, provocando la morte delle cellule nervose, in modo simile a quanto avviene durante un infarto miocardico, ma a livello delle vie visive.
Come si manifesta?
La neuropatia ottica ischemica anteriore (NOIA) si manifesta con una riduzione improvvisa della vista, spesso in una parte specifica del campo visivo, centrale, superiore o inferiore (difetto altitudinale). Nei casi di difetto altitudinale, guardando in orizzontale il paziente può non vedere più il pavimento o il soffitto della stanza, mentre in alcuni casi la visione centrale rimane intatta. Nella maggior parte dei casi, il calo visivo interessa un occhio e viene notato al risveglio. La NOIA è la causa più comune di neuropatia ottica acuta nei pazienti con più di 50 anni.
Quanti tipi di NOIA esistono?
La neuropatia ottica ischemica si può differenziare in due forme:
la forma non arteritica
la forma arteritica
Quali sono le cause per la forma non arteritica?
La neuropatia ottica ischemica anteriore non arteritica è causata da un’occlusione delle arterie ciliari posteriori brevi su base aterosclerotica, associata a fattori di rischio sia generali sia locali. Tra i fattori generali rientrano quelli tipici delle patologie ischemiche, come ipertensione arteriosa, diabete mellito, fumo, colesterolo alto, iperomocisteinemia, obesità e scarsa attività fisica. Tra i fattori locali si segnala la conformazione del disco ottico, piccolo e “affollato” di vasi sanguigni. Se i fattori di rischio sistemici non vengono trattati, la patologia può diventare bilaterale, con coinvolgimento dell’altro occhio nel 25-50% dei casi entro cinque anni.
Quali sono le cause della forma arteritica?
La forma arteritica è causata dall’occlusione delle arterie ciliari posteriori brevi a seguito di una malattia infiammatoria sistemica dei piccoli vasi, nota come arterite di Horton o arterite a cellule giganti. Se non riconosciuta e trattata tempestivamente,può interessare rapidamente anche l’occhio controlaterale. La diagnosi iniziale si basa sull’aumento dei valori plasmatici di VES e PCR, mentre la conferma definitiva viene ottenuta tramite biopsia dell’arteria temporale.
Come si esegue la diagnosi?
La diagnosi di neuropatia ottica ischemica anteriore (NOIA) si basa sui sintomi riferiti dal paziente, sull’esame del fondo oculare, sul campo visivo e sulla fluorangiografia. In fase acuta, il disco ottico appare edematoso e biancastro, spesso accompagnato da emorragie a fiamma e noduli cotonosi peripapillari, generalmente localizzati in una porzione del disco. Dopo alcune settimane l’edema si risolve e compare pallore del disco ottico, diffuso o localizzato, dovuto all’atrofia delle fibre nervose. Il campo visivo può evidenziare un caratteristico difetto altitudinale o uno scotoma centrale. La fluorangiografia mostra spesso un ritardo di riempimento delle arterie ciliari posteriori brevi e un leakage tardivo della papilla ottica, utile per confermare la diagnosi e valutare i deficit di perfusione del nervo ottico, fondamentale anche per comprendere l’efficacia della somministrazione di eventuali farmaci. Nella forma arteritica, è importante valutare i valori ematochimici come emocromo, VES e PCR, che risultano alterati.
Come si cura?
La forma non arteritica, in fase acuta, può essere trattata con la somministrazione di steroidi per via orale, che servono a ridurre l’edema del disco ottico. Di notevole importanza è la prevenzione della neuropatia ischemica nell’occhio sano. In questi casi si raccomanda l’uso di antiaggreganti piastrinici e il controllo dei fattori di rischio cardiovascolari come l’ipertensione arteriosa sistemica, il diabete e le dislipidemie (variazioni e aumenti dei grassi e lipidi nel sangue). La forma arteritica viene trattata in acuto con boli (steroidi) per via endovenosa e poi per via orale con terapia a scalare. Per la gestione del paziente è importante la collaborazione del medico internista e del medico immunologo.
Effetti sulla vita quotidiana
La perdita visiva causata dalla neuropatia ottica ischemica può rendere difficili molte attività quotidiane, come leggere, guidare o svolgere attività domestiche. Per affrontare queste situazioni, è fondamentale non solo accettare le limitazioni, ma anche ricorrere a interventi di riabilitazione visiva. La riabilitazione visiva mira a ottimizzare le capacità visive residue dei pazienti e a sviluppare strategie pratiche per migliorarne l’autonomia. Tra gli strumenti utilizzati ci sono, ad esempio, dispositivi digitali con funzione di lettura vocale e software di ingrandimento dello schermo; anche modifiche ambientali, come un’illuminazione adeguata e una disposizione funzionale degli spazi domestici, possono favorire la sicurezza e l’indipendenza. Un percorso di riabilitazione visiva personalizzato, seguito da professionisti specializzati, permette di mantenere un buon livello di autonomia e di adattarsi più facilmente alle nuove esigenze visive, migliorando così la qualità della vita quotidiana.
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus.
Contatta l’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Il Numero Verde di consultazione oculistica è attivo dal lunedì al venerdì dalle 10 alle 13. Scrivi nel Forum: un medico oculista ti risponderà gratuitamente.
La neurite ottica è una patologia del nervo ottico che comprende quadri clinici simili a livello di segni e sintomi, ma molto diversi come eziologia. Non sempre, infatti, le cause di questa malattia oculare sono di natura infiammatoria, ma anche demielinizzante, degenerativa o tossica.
Nei pazienti ultracinquantenni si presenta spesso come neuropatia ottica ischemica, nella forma arteritica o non arteritica: queste due forme si differenziano tra loro principalmente per i valori della VES, acronimo che sta ad indicare la velocità di eritrosedimentazione ossia di sedimentazione dei globuli rossi che, nel primo caso, risultano molto più elevati.
Sia la forma arteritica che quella non arteritica sono caratterizzate da una perdita della visione repentina e, purtroppo, spesso molto invalidante. Inoltre, nella prima forma spesso possono coesistere sintomi come mal di testa, fastidio o dolore del cuoio capelluto, dolore alla masticazione o alle articolazioni, assieme a segni come la palpabilità dell’arteria temporale.
Che sintomi dà?
Una perdita rapida delle funzioni visive che può essere lieve o, più spesso, grave. I colori appaiono sbiaditi. Sono usuali dolori oculari nella forma retrobulbare, soprattutto nei movimenti di sguardo. Si riscontrano alterazioni, di solito specifiche e che aiutano nella diagnosi, del campo visivo.
Che cause ha?
La causa della neuropatia ottica ischemica è fondamentalmente di natura vascolare.
Ad una stimolazione fatta con una luce si riscontra un rallentato riflesso della pupilla (difetto pupillare relativo afferente); gli esami del campo visivo – sia manuale che computerizzato – confermeranno la presenza di aree di non percezione visiva (scotomi). L’esame PEV (potenziali evocati visivi) mostrerà alterazioni nella trasmissione del segnale lungo il nervo ottico.
Come si fa la diagnosi?
All’esame del fondo oculare il medico oculista può riscontrare la testa del nervo ottico (papilla) rigonfia associata ad emorragie circostanti; ma la papilla può essere normale nel caso in cui si tratti di una neurite ottica retrobulbare.
Necessario per la diagnosi è, comunque, l’esame del campo visivo. Per le neuriti ottiche sono utili indagini diagnostiche quali la fluorangiografia e i potenziali evocati visivi (PEV).
È importante comunque sottolineare ancora una volta che la neurite ottica può essere un sintomo iniziale di diverse condizioni patologiche. Pertanto un esame clinico-medico completo può aiutare ad escludere eventuali malattie correlate.
Gli esami ematochimici consentono di ricercare la presenza di parametri infiammatori, come la VES o la proteina C reattiva. Una velocità di eritrosedimentazione (VES) elevata può aiutare a determinare se la neurite ottica è causata dall’infiammazione delle arterie craniche (arterite temporale); inoltre, le analisi del sangue consentono di riscontrare la presenza di anticorpi anti-mielina (per indagare su malattie autoimmuni) e segni di eventuali infezioni virali e batteriche.
Chi avesse avuto un primo episodio di neurite ottica in genere verrà sottoposto a una risonanza magnetica per cercare eventuali lesioni a carico del sistema nervoso centrale. Questo test di imaging consente, infatti, di determinare se la mielina sia stata danneggiata e può aiutare ad eseguire un’eventuale diagnosi di sclerosi multipla (soprattutto nelle giovani donne, purtroppo più predisposte alla patologia), dimostrando la presenza di anomalie caratteristiche di questa patologia.
Qual è la terapia?
Nella neuropatia ottica ischemica arteritica e in quella retrobulbare si impiegano steroidi per via sistemica, inizialmente per via endovenosa. Rimane fondamentale una diagnosi precoce perché il trattamento andrebbe effettuato il prima possibile per cercare di limitare i danni. Basti pensare anche che, nella forma arteritica, caratterizzata spesso da un recupero visivo scarso, una terapia adeguata riduce fortemente il rischio anche di un episodio analogo nell’occhio sano.
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus Leggi le condizioni generali di consultazione di questo sito
Pagina pubblicata il 1 luglio 2008. Ultimo aggiornamento: 11 febbraio 2019.
Ultima revisione scientifica: 22 novembre 2022
Contatta l’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Il Numero Verde di consultazione oculistica è attivo dal lunedì al venerdì dalle 10 alle 13. Scrivi nel Forum: un medico oculista ti risponderà gratuitamente.
La retinopatia ipertensiva è una malattia oculare che si riscontra in soggetti che presentano una pressione arteriosa elevata. L’ipertensione arteriosa sistemica è una patologia di frequente riscontro che, se diagnosticata e curata in tempo, non provoca danni irreversibili. In caso contrario può portare a tutta una serie di problematiche a livello di vari organi, quali il cervello, il cuore, il fegato, il rene e l’occhio. La retinopatia ipertensiva in fase iniziale può risultare asintomatica e non comportare particolari disturbi visivi per il paziente, nei casi più gravi e avanzati invece, si rileva un netto peggioramento della visione perché viene alterato il corretto funzionamento della retina a causa di un ampio spettro di alterazioni vascolari.
Da cosa è causata?
Come anticipato in precedenza, la causa di questa patologia è la pressione arteriosa troppo alta. Infatti, chi ha una pressione cronicamente oltre i valori normali, a lungo andare tende a soffrire di alterazioni dei vasi retinici ossia:
restringimento del lume vasale ed aumento della tortuosità del vaso stesso (si tratta di un meccanismo di compenso da parte dell’occhio per proteggere la retina);
vasodilatazione con conseguente stravaso di liquido nel tessuto retinico (essudati cotonosi) e, in un secondo momento, comparsa di emorragie;
comparsa di edema retinico, con formazione di essudati duri intorno alla fovea.
In base a tali meccanismi evolutivi, la retinopatia ipertensiva può essere suddivisa clinicamente in quattro stadi:
Stadio 1: è caratterizzato da un lieve e diffuso restringimento arteriolare con comparsa di tortuosità dei vasi retinici.
Stadio 2: comparsa di schiacciamenti artero-venosi: la vena subisce uno spostamento repentino dopo l’incrocio artero-venoso, oppure si crea un ingorgo ematico che rende la vena più grossa e tortuosa prima dell’incrocio e più sottile e lineare dopo di esso. Le arterie retiniche appaiono ristrette, rigide, rettilinee con calibro irregolare ed accentuazione del loro riflesso (definito a “filo di rame”, più tardivamente a “filo d’argento”).
Stadio 3: presenza di edema retinico, emorragie a fiamma, essudati cotonosi, che si presentano come chiazze biancastre superficiali, essudati duri, che si localizzano principalmente a livello della macula. Quando l’edema e gli essudati interessano la macula si parla di “stella maculare”, condizione associata ad una grave compromissione della vista.
Stadio 4: comparsa di edema della testa del nervo ottico (papilla da stasi) per la concomitante presenza di ipertensione endocranica, con quadro clinico molto grave.
Quali sono i sintomi?
Nelle forme lievi non sono presenti disturbi, mentre nelle forme avanzate di retinopatia ipertensiva la visione può risultare annebbiata, le immagini apparire distorte, con calo del visus importante e comparsa di disturbi neurologici.
Come si effettua la diagnosi?
La diagnosi avviene attraverso l’esame del fondo oculare, previa instillazione di un collirio per la dilatazione delle pupille (midriasi). È possibile, in questo modo, valutare dimensione e decorso dei vasi sanguigni, eventuale presenza di emorragie, di essudati o di edema maculare. A seconda del grado di alterazione arterioso e venoso nonché della presenza di lesioni (comprese eventuali zone ischemiche) si determina lo stadio della malattia.
Un’eventuale fluorangiografia(con cui si inietta per via endovenosa una sostanza fluorescente e si eseguono delle fotografie della retina in tempi diversi)può essere utile per evidenziare alterazioni precoci dei vasi retinici e studiare l’evoluzione della patologia.
Qual è la terapia?
La terapia si basa esclusivamente sul controllo, attraverso opportune cure, dell’ipertensione arteriosa sistemica (pressione alta). È importante, quindi, l’esame semestrale del fondo oculare dall’oculista in chi è iperteso, anche per verificare il grado di efficacia della terapia e valutare lo stadio della malattia retinica. Può accadere che si scopra di essere ipertesi soltanto dopo una visita oculistica, quando lo specialista rileva la presenza dei segni iniziali della retinopatia ipertensiva.
Prendo regolarmente un medicinale che riporta la mia pressione a valori normali, posso stare tranquillo/a?
Mediante il controllo del fondo oculare si possono evidenziare le alterazioni retiniche a carico del microcircolo, contribuendo così alla diagnosi precoce di uno stato ipertensivo latente e consentendo – con l’ausilio del cardiologo o del medico di base – un trattamento farmacologico adeguato. In questo modo si potrà formulare un giudizio sull’efficacia della terapia antipertensiva in atto. È evidente, quindi, che un controllo periodico della condizione del fondo dell’occhio potrà fornire un’informazione accurata sull’evoluzione di eventuali alterazioni retiniche causate dall’ipertensione.
La mia pressione è poco più alta della norma, come devo comportarmi?
La pressione arteriosa al di sopra della norma, in particolare quella diastolica (la minima), provoca danni ai vasi sanguigni degli occhi. Più a lungo e più è alta la pressione arteriosa, più rischia di essere grave il danno a livello retinico. È consigliato quindi, in questi casi, farsi seguire da un cardiologo o da un internista, i quali hanno le competenze più adeguate. Un’opportuna terapia farmacologica, associata a una dieta corretta (poco sale e pochi grassi), oltre a uno stile di vita meno sedentario, praticare regolarmente un’attività fisica moderata, possono portare a un miglioramento del quadro clinico.
Nota: la pressione alta degli occhi (ipertensione oculare) è soltanto marginalmente legata a quella sanguigna: difficilmente chi soffre di pressione arteriosa elevata ha valori elevati di quella oculare (o viceversa). Tuttavia recenti studi epidemiologici hanno confermato l’esistenza di una correlazione, tanto che chi soffre di pressione arteriosa alta sarebbe anche più a rischio di essere colpito da glaucoma.
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus Leggi le condizioni generali di consultazione di questo sito
Pagina pubblicata il 31 gennaio 2013. Ultimo aggiornamento: 26 aprile 2023.
Ultima revisione scientifica: 26 aprile 2023.
Contatta l’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Il Numero Verde di consultazione oculistica è attivo dal lunedì al venerdì dalle 10 alle 13. Scrivi nel Forum: un medico oculista ti risponderà gratuitamente.
La malattia di Best, conosciuta anche come distrofia maculare vitelliforme, è una condizione ereditaria che provoca una degenerazione della retina, in particolare della macula, ossia la zona centrale responsabile della visione dettagliata e della percezione dei colori. Sebbene la diagnosi di questa malattia avvenga di solito durante l’adolescenza, il deterioramento della vista si manifesta in genere in età adulta. La progressione della malattia è variabile, ma la visione periferica tende a rimanere intatta.
Da cosa è causata?
La malattia di Best è una condizione ereditaria che segue un pattern di trasmissione autosomica dominante, il che significa che una persona con una copia mutata del gene ha una probabilità del 50% di trasmettere la malattia ai figli. È causata da mutazioni del gene VMD2, situato sul cromosoma 11q13, che è responsabile del trasporto degli acidi grassi polinsaturi. Quando il gene è mutato, si verifica un accumulo di lipofuscina nell’epitelio pigmentato retinico, un fenomeno che contribuisce allo sviluppo della malattia. I maschi sono colpiti più spesso rispetto alle femmine (3:1).
Quali sono i sintomi?
Alla nascita, i bambini affetti presentano una visione normale. Tuttavia, con il progredire della malattia, si sviluppano diverse fasi che interessano principalmente la macula, la parte centrale della retina responsabile della visione dettagliata. La progressione della malattia è caratterizzata da stadi distinti, ognuno dei quali presenta segni e sintomi specifici.
Nella fase iniziale, detta stadio previtelliforme (stadio 1), non si osservano sintomi evidenti. Il secondo stadio (detto vitelliforme) è contraddistinto dalla formazione di una lesione giallastra, simile a un tuorlo d’uovo, sulla macula. In questa fase, i difetti visivi sono generalmente lievi. Successivamente, il materiale che compone la lesione assume un aspetto meno omogeneo e può apparire come un “uovo strapazzato”. Al terzo stadio, la lesione si trasforma in una sostanza liquida giallastra, che può rompersi, provocando danni alle cellule retiniche sottostanti. Questo stadio comporta la comparsa di disturbi visivi come le metamorfopsie, in cui le linee rette vengono percepite come ondulate, o difficoltà nella lettura dei caratteri più piccoli. Infine, nell’ultimo stadio (stadio IV), il materiale giallastro che ha causato le lesioni tende a ritirarsi e a scomparire, lasciando cicatrici e cellule danneggiate. Il tentativo di riparazione da parte dell’occhio può comportare la formazione di nuovi vasi sanguigni, spesso difettosi e incapaci di sostenere una corretta circolazione. Questi vasi possono sanguinare, portando alla formazione di tessuti cicatriziali e al deterioramento della visione centrale. Nel corso della malattia, i pazienti possono sviluppare difficoltà nella discriminazione dei colori e una compromissione della visione centrale, ma la visione periferica e l’adattamento al buio rimangono generalmente intatti. In alcuni casi, i pazienti possono rimanere asintomatici per lungo tempo. La gravità e il momento in cui la vista si deteriora variano significativamente da persona a persona, rendendo difficile prevedere l’evoluzione della malattia. Sebbene i primi stadi si manifestino tipicamente nell’infanzia o nell’adolescenza senza particolari problematiche, la compromissione visiva significativa tende a verificarsi negli stadi avanzati, solitamente dalla quarta decade di vita in poi.
Come si effettua la diagnosi?
La diagnosi della malattia di Best si basa su un’accurata valutazione clinica che include l’anamnesi familiare, l’osservazione delle caratteristiche tipiche delle lesioni sulla macula e l’esecuzione di test specialistici. La condizione è solitamente identificata durante l’infanzia o l’adolescenza.
Esame oftalmologico
Il primo passo per diagnosticare la malattia è un esame oftalmologico completo, che include la valutazione dell’acuità visiva e un’analisi approfondita del fondo oculare a pupilla dilatata, finalizzato a identificare eventuali lesioni vitelliformi, atrofia o cicatrici nella macula.
La tomografia a coerenza ottica (OCT).
È un’importante tecnica di imaging che permette di visualizzare i depositi sottoretinici, aiutando non solo a monitorare la progressione della malattia, ma anche a stadiarla nel tempo.
L’autofluorescenza.
Insieme alla fotografia del fondo oculare viene utilizzata per documentare l’evoluzione delle lesioni e la loro distribuzione nella retina.
La fluorangiografia (FAG)
Utile per visualizzare la circolazione della retina ed identificare la presenza di lesioni.
Poiché la malattia di Best è solitamente bilaterale, ma spesso presenta una distribuzione asimmetrica, entrambi gli occhi vengono esaminati per identificare eventuali alterazioni. È fondamentale anche escludere altre patologie maculari in base all’età del paziente, alla tipica morfologia delle lesioni e ai risultati dei test oftalmologici.
Elettrofisiologia e genetica
Per confermare ulteriormente la diagnosi vengono effettuati test elettrofisiologici e genetici. L’elettrooculogramma (EOG) solitamente rivela una riduzione significativa della risposta alla luce, mentre l’elettroretinogramma (ERG) tende a restare relativamente conservato. I test genetici sono fondamentali per identificare varianti della malattia e per distinguerla da altre distrofie maculari ereditarie, offrendo anche informazioni cruciali per la prognosi a lungo termine del paziente.
Inoltre, lo screening genetico familiare è un passo importante, poiché consente di identificare eventuali portatori asintomatici e orientare le future strategie di monitoraggio e prevenzione per i membri della famiglia.
Quali terapie sono disponibili?
Al momento, purtroppo, non esiste una cura definitiva per la malattia di Best. I trattamenti disponibili si concentrano principalmente sul miglioramento dei sintomi e della qualità di vita. Ad esempio, alcune vitamine specifiche potrebbero rallentare la progressione della malattia, come suggeriscono alcuni studi. Inoltre, un’adeguata correzione con occhiali o lenti a contatto, può aiutare a gestire i problemi visivi, migliorando la vita quotidiana. Esistono anche terapie basate sulla riabilitazione visiva e l’utilizzo di dispositivi speciali che possono aiutare i pazienti a sfruttare al meglio il loro residuo visivo. Per le complicanze, come la formazione di neovascolarizzazioni, si possono usare trattamenti come il laser o le iniezioni intravitreali di farmaci anti-angiogenici (anti-VEGF) che servono a rallentare la progressione della malattia. È importante che chi soffre di questa condizione faccia regolari controlli con un oculista per monitorare l’andamento della malattia e intervenire prontamente se necessario.
Infine, la terapia genica è una speranza per il futuro. Ci sono attualmente in corso numerosi studi, che potrebbero offrire nuove soluzioni terapeutiche per trattare la malattia di Best.
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus Leggi le condizioni generali di consultazione di questo sito
Ultima revisione scientifica: 23 aprile 2026.
Contatta l’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Il Numero Verde di consultazione oculistica è attivo dal lunedì al venerdì dalle 10 alle 13. Scrivi nel Forum: un medico oculista ti risponderà gratuitamente.


 Lo “scotoma” (dal greco skótos, “oscurità”) è un difetto lacunare del campo visivo. E’ correlato a una riduzione della sensibilità retinica (scotoma relativo) o a una scomparsa completa della sensibilità stessa (scotoma assoluto) in alcune aree della retina, dove l’immagine non risulta più percepibile oppure se ne ha una percezione sbiadita.
Lo “scotoma” (dal greco skótos, “oscurità”) è un difetto lacunare del campo visivo. E’ correlato a una riduzione della sensibilità retinica (scotoma relativo) o a una scomparsa completa della sensibilità stessa (scotoma assoluto) in alcune aree della retina, dove l’immagine non risulta più percepibile oppure se ne ha una percezione sbiadita.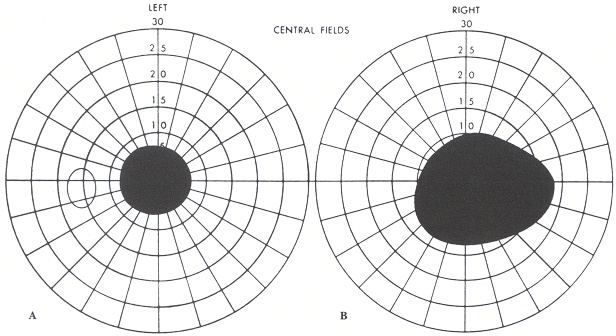 In linea di massima lo scotoma non comporta disturbi importanti (a meno che non sia di dimensioni notevoli o localizzato nella zona centrale), tanto che in alcune patologie (tipo il glaucoma) ci si rende conto della sua presenza solo quando il campo visivo è già danneggiato e si è, quindi, ristretto. Per scongiurare quest’eventualità è consigliabile sottoporsi periodicamente a un controllo oculistico.
In linea di massima lo scotoma non comporta disturbi importanti (a meno che non sia di dimensioni notevoli o localizzato nella zona centrale), tanto che in alcune patologie (tipo il glaucoma) ci si rende conto della sua presenza solo quando il campo visivo è già danneggiato e si è, quindi, ristretto. Per scongiurare quest’eventualità è consigliabile sottoporsi periodicamente a un controllo oculistico.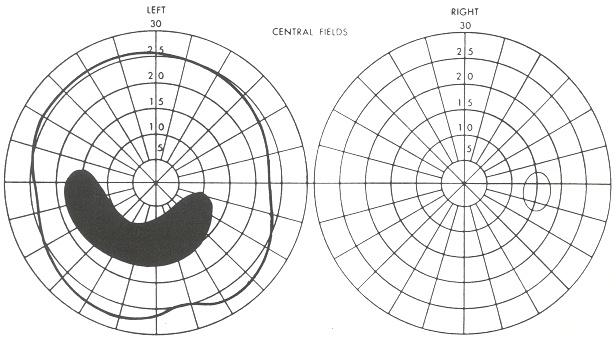 Si può classificare in:
Si può classificare in:

 L’esordio della malattia è spesso subdolo ed i sintomi sono generalmente monolaterali. I disturbi visivi più frequentemente rilevati sono:
L’esordio della malattia è spesso subdolo ed i sintomi sono generalmente monolaterali. I disturbi visivi più frequentemente rilevati sono:
 Attraverso l’esame del
Attraverso l’esame del 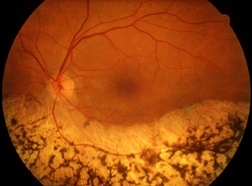

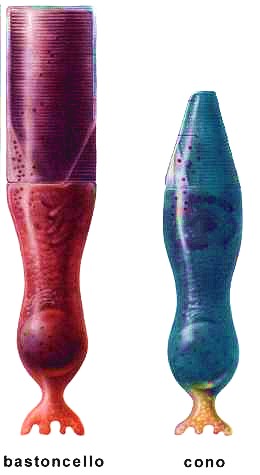




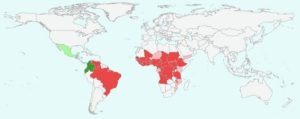
 La neurite ottica è una patologia del
La neurite ottica è una patologia del  La retinopatia ipertensiva è una malattia oculare che si riscontra in soggetti che presentano una pressione arteriosa elevata. L’ipertensione arteriosa sistemica è una patologia di frequente riscontro che, se diagnosticata e curata in tempo, non provoca danni irreversibili. In caso contrario può portare a tutta una serie di problematiche a livello di vari organi, quali il cervello, il cuore, il fegato, il rene e l’occhio. La retinopatia ipertensiva in fase iniziale può risultare asintomatica e non comportare particolari disturbi visivi per il paziente, nei casi più gravi e avanzati invece, si rileva un netto peggioramento della visione perché viene alterato il corretto funzionamento della retina a causa di un ampio spettro di alterazioni vascolari.
La retinopatia ipertensiva è una malattia oculare che si riscontra in soggetti che presentano una pressione arteriosa elevata. L’ipertensione arteriosa sistemica è una patologia di frequente riscontro che, se diagnosticata e curata in tempo, non provoca danni irreversibili. In caso contrario può portare a tutta una serie di problematiche a livello di vari organi, quali il cervello, il cuore, il fegato, il rene e l’occhio. La retinopatia ipertensiva in fase iniziale può risultare asintomatica e non comportare particolari disturbi visivi per il paziente, nei casi più gravi e avanzati invece, si rileva un netto peggioramento della visione perché viene alterato il corretto funzionamento della retina a causa di un ampio spettro di alterazioni vascolari.