

Retinopatia sierosa centrale
Cos’è?
Si tratta di una malattia oculare caratterizzata dal sollevamento della zona centrale della retina a causa dell’accumulo di liquido sieroso.
In particolare, si evidenzia il distacco delimitato di uno strato retinico intermedio (il neuroepitelio) nella regione maculare, a volte associato a distacco sieroso dell’epitelio pigmentato retinico (EPR). Lo stravaso di siero, dai vasi coroideali, passando attraverso lesioni focali dell’EPR (punti di fuga), si accumula nello spazio sottoretinico causandone il distacco. Si tratta di una patologia sporadica, generalmente monolaterale, che può tuttavia colpire entrambi gli occhi in maniera più o meno simmetrica. In genere il sesso maschile tra i 30 e i 50 anni di età (con tendenza ad una personalità di tipo ansioso) è quello più colpito; le donne affette da CSC hanno generalmente un’età media più alta degli uomini. È meno frequente nella razza nera e può presentarsi in forme particolarmente gravi negli asiatici.

Che cause ha?
Sulle cause che determinano la sierosa centrale non si hanno dati certi. Un elemento significativo, che accomuna quasi tutti coloro che ne sono affetti, è che sembrerebbe essere favorita dello stress (si presenta di solito in persone particolarmente attive e competitive). L’induzione o l’aggravamento della malattia sono stati associati a fattori predisponenti quali: ipertensione arteriosa, trapianto di organi, reflusso gastroesofageo, abuso di alcool, terapia steroidea orale e/o inalatoria, gravidanza e patologie sistemiche come il Lupus Eritematoso Sistemico e la Sindrome di Cushing.
Sono stati osservati anche importanti fattori genetici che possono predisporre allo sviluppo della CSC. [van Dijk EHC, Schellevis RL, Breukink MB, Mohabati D, Dijkman G, Keunen JEE, Yzer S, den Hollander AI, Hoyng CB, de Jong EK, Boon CJF, “[Familial Central Serous Chorioretinopathy”, Retina, 2017 Nov 28. doi: 10.1097/IAE.0000000000001966 (Epub ahead of print)].
Quali sono i sintomi e i segni della CSC?
 L’esordio della malattia è spesso subdolo ed i sintomi sono generalmente monolaterali. I disturbi visivi più frequentemente rilevati sono:
L’esordio della malattia è spesso subdolo ed i sintomi sono generalmente monolaterali. I disturbi visivi più frequentemente rilevati sono:
- visione appannata (spesso si ha l’impressione di guardare come attraverso una goccia d’acqua);
- presenza di uno scotoma centrale relativo;
- metamorfopsie (distorsione delle immagini);
- visione più scura;
- colori sbiaditi;
- riduzione dell’acuità visiva.
All’esame del fondo oculare si osserva tipicamente un sollevamento sieroso della retina neurosensoriale localizzato nella regione maculare. Si possono evidenziare uno o più distacchi sierosi dell’epitelio pigmentato, in associazione o anche in assenza del distacco retinico. Col tempo, in sede sottoretinica, si possono notare piccoli precipitati giallastri di materiale proteico o di derivazione dai segmenti esterni dei fotorecettori nell’area del distacco
Come si effettua una diagnosi?
Per identificare la presenza di una corioretinopatia sierosa centrale è indispensabile sottoporsi ad una visita oculistica completa. In prima istanza, lo specialista attraverso l’anamnesi raccoglie più informazioni possibili sul paziente: età, stile di vita, farmaci assunti, presenza di eventuali patologie sistemiche, modalità e tempi d’insorgenza dei disturbi visivi. Alla visita oculistica l’esame del visus può risultare più o meno alterato, il segmento anteriore e la pressione oculare sono di solito nella norma, all’esame del fondo oculare si evidenziano, in sede maculare, i segni tipici della malattia (vedi paragrafo precedente). Per un approfondimento può essere utile sottoporsi ad esami strumentali specifici.
- L’OCT (tomografia a coerenza ottica), è una tecnica diagnostica non invasiva che consente di rilevare e monitorare nel tempo, il sollevamento retinico maculare nei pazienti con CSC. Nelle forme croniche, all’esame OCT è possibile rilevare la presenza di cisti intraretiniche e un ispessimento coroideale. In caso di complicanze, si può evidenziare la comparsa di neovasi, facilmente individuabili con l’angio-OCT, una metodica angiografica semplice e non invasiva, che non ha bisogno dell’ iniezione in vena di coloranti.
- La fluorangiografia (FAG), è l’esame che da sempre è stato utilizzato per definire le caratteristiche cliniche della CSC. E’ tipico il rilievo di un “leakage” focale o multifocale a livello dell’epitelio pigmentato retinico. Gli eventuali piccoli distacchi dell’epitelio pigmentato risultano iperfluorescenti e chiaramente rilevabili. Nella CSC cronica la fluorangiografia può evidenziare la presenza di un’ epiteliopatia retinica diffusa.
- L’ angiografia con verde di indocianina, consente di acquisire informazioni importanti per monitorare il decorso della malattia. Con l’esame è possibile rilevare la diffusione del colorante in aree più o meno estese della coroide sia in corrispondenza delle alterazioni dell’epitelio pigmentato evidenziate dalla fluorangiografia, che in zone dove l’epitelio appare integro. Un’importante iperpermeabilità di alcune zone della coroide caratterizza i pazienti con CSC, sia nei momenti di attività che di inattività della malattia. L’angiografia con verde di indocianina permette anche di individuare eventuali neovascolarizzazioni occulte che possono simulare o complicare una CSC cronica.
- L’autofluorescenza è un esame semplice per valutare nel dettaglio il fondo oculare: consente di evidenziare le aree di degenerazione e atrofia dell’epitelio pigmentato (e dei fotorecettori) come conseguenza della prolungata persistenza di liquido sotto la retina. Rappresenta, pertanto, una metodica importante per riconoscere la cronicità della malattia. Da una semplice immagine in autofluorescenza possiamo renderci conto se siamo di fronte a un processo essudativo recente, oppure che dura da diverso tempo (mesi o anni).
Il liquido che si è accumulato tra gli strati retinici può riassorbirsi spontaneamente entro alcuni mesi e si può recuperare l’acuità visiva originaria; ma è comunque importante farsi seguire da uno specialista che possa prescrivere gli esami più opportuni, monitorando costantemente lo stato di salute della retina. Inoltre sono stati rilevati cambiamenti nel microcircolo retinico di persone cronicamente affette da corioretinopatia sierosa centrale. [Sugiura A, Fujino R, Takemiya N, Shimizu K, Matsuura M, Murata H, Inoue T, Obata R, Asaoka R, “[The association between visual function and retinal structure in chronic central serous chorioretinopathy“, Sci Rep. 2017 Nov 24;7(1):16288. doi: 10.1038/s41598-017-16339-9].
C’è una terapia univoca?
No, non vi è un protocollo terapeutico specifico, perché nessun trattamento medico sino ad oggi si è dimostrato sempre valido. Si consiglia generalmente di ridurre lo stress e di condurre uno stile di vita più tranquillo ma queste, chiaramente, sono indicazioni che non sempre i pazienti riescono a seguire. Svariati tipi di farmaci (tranquillanti, antinfiammatori non steroidei, beta bloccanti) sono stati provati senza grande successo, così come inefficace ed anzi in grado di indurre un peggioramento clinico, si è dimostrato essere il cortisone. In qualche caso sembrerebbe avere una certa utilità l’uso di diuretici che riducendo l’accumulo di fluidi sotto la retina, possono accelerare la risoluzione dei sintomi. Attualmente la letteratura medico-scientifica riporta buoni risultati ottenuti con la fotocoagulazione laser, che andrebbe ad agire proprio sul punto di perdita; ma questa eventualità, data la buona prognosi, deve essere valutata dal singolo oculista con molta attenzione e in casi particolari. Il trattamento laser non è indicato, invece, nei casi in cui il punto di fuga sia foveale o nei casi di punti di fuga multipli. Dobbiamo inoltre ricordare che la fotocoagulazione comporta la distruzione di tessuto retinico con eventuali danni a livello visivo (calo del visus, scotoma localizzato, riduzione della sensibilità al contrasto, alterata percezione dei colori). Esiste anche un’altra opzione terapeutica rappresentata dalla terapia fotodinamica indicata anche in quei casi in cui i punti di fuga si presentino iuxtafoveali o sottofoveali e nelle forme croniche; seppur valida, la terapia fotodinamica non è comunque esente da possibili effetti collaterali importanti quali per esempio l’atrofia dell’EPR e la formazione di scotomi localizzati. Più di recente è stato proposto un trattamento con laser giallo micropulsato, che dovrebbe attivare l’epitelio pigmentato favorendo il riassorbimento del liquido sottoretinico. L’efficacia di questo tipo di laser è variabile da un caso all’altro, risulta comunque essere un trattamento indolore, sicuro e potenzialmente ripetibile. Infine, nei casi in cui la CSC si complichi con una neovascolarizzazione coroideale, è indicato il trattamento con iniezioni intravitreali di farmaci anti-VEGF.
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Leggi le condizioni generali di consultazione di questo sito
Pagina pubblicata il 11 marzo 2008. Ultimo aggiornamento: 13 febbraio 2025
Ultima revisione scientifica: 13 febbraio 2025


 Attraverso l’esame del
Attraverso l’esame del 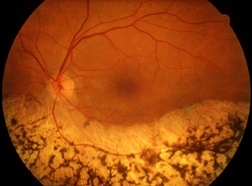





 La neurite ottica è una patologia del
La neurite ottica è una patologia del  La retinopatia ipertensiva è una malattia oculare che si riscontra in soggetti che presentano una pressione arteriosa elevata. L’ipertensione arteriosa sistemica è una patologia di frequente riscontro che, se diagnosticata e curata in tempo, non provoca danni irreversibili. In caso contrario può portare a tutta una serie di problematiche a livello di vari organi, quali il cervello, il cuore, il fegato, il rene e l’occhio. La retinopatia ipertensiva in fase iniziale può risultare asintomatica e non comportare particolari disturbi visivi per il paziente, nei casi più gravi e avanzati invece, si rileva un netto peggioramento della visione perché viene alterato il corretto funzionamento della retina a causa di un ampio spettro di alterazioni vascolari.
La retinopatia ipertensiva è una malattia oculare che si riscontra in soggetti che presentano una pressione arteriosa elevata. L’ipertensione arteriosa sistemica è una patologia di frequente riscontro che, se diagnosticata e curata in tempo, non provoca danni irreversibili. In caso contrario può portare a tutta una serie di problematiche a livello di vari organi, quali il cervello, il cuore, il fegato, il rene e l’occhio. La retinopatia ipertensiva in fase iniziale può risultare asintomatica e non comportare particolari disturbi visivi per il paziente, nei casi più gravi e avanzati invece, si rileva un netto peggioramento della visione perché viene alterato il corretto funzionamento della retina a causa di un ampio spettro di alterazioni vascolari.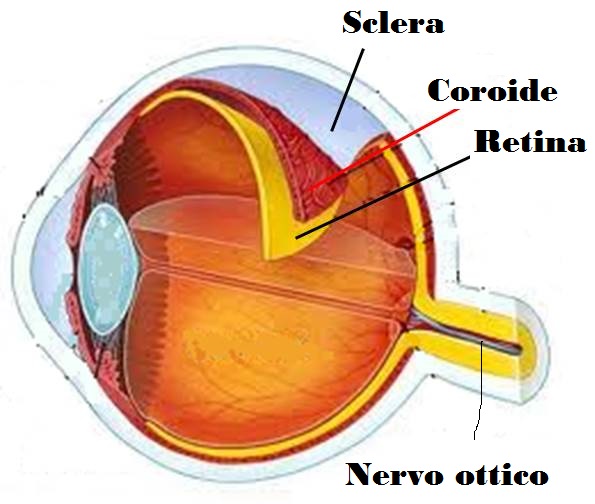
 L’occhio umano, come tutte le altre strutture dell’organismo che sono in contatto con l’ambiente esterno, è spesso soggetto all’attacco di microrganismi ad azione patogena (batteri, virus, protozoi, ecc.), responsabili d’infezioni più o meno importanti ed estese. In tal senso risulta fondamentale l’azione protettiva contro le minacce ambientali (agenti esogeni pericolosi), svolta normalmente dalle palpebre e dal film lacrimale.
L’occhio umano, come tutte le altre strutture dell’organismo che sono in contatto con l’ambiente esterno, è spesso soggetto all’attacco di microrganismi ad azione patogena (batteri, virus, protozoi, ecc.), responsabili d’infezioni più o meno importanti ed estese. In tal senso risulta fondamentale l’azione protettiva contro le minacce ambientali (agenti esogeni pericolosi), svolta normalmente dalle palpebre e dal film lacrimale.