

Retinite pigmentosa
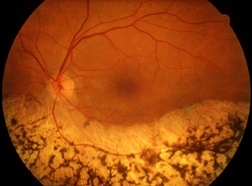
Si tratta di una patologia rara che appartiene a un gruppo di malattie ereditarie caratterizzate da una degenerazione progressiva della retina in entrambi gli occhi. Provoca la perdita graduale della visione notturna e del campo visivo periferico, ma agli ultimi stadi si può verificare anche una perdita della visione centrale. A causa della retinite pigmentosa (nota anche come “retinosi pigmentaria” o “retinopatia pigmentosa”) si verifica una perdita dell’acutezza visiva, con un progressivo restringimento del campo visivo, che può progredire fino all’ipovisione e, nei casi più gravi, alla cecità.
Come funziona la retina?
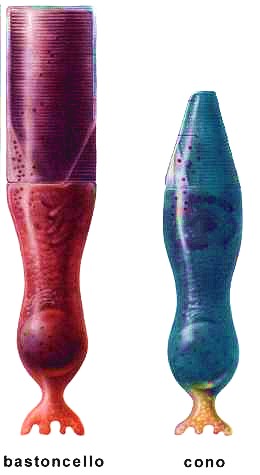
La retina è costituita da cellule nervose disposte in strati; immaginiamola come una pellicola fotografica. È in assoluto la struttura del nostro occhio più importante ed è essenziale nel processo della visione. Infatti i raggi luminosi che giungono dall’esterno vengono messi a fuoco sulla retina e qui trasformati in segnali elettrochimici che, attraverso il nervo ottico, giungono al cervello, che normalmente le percepisce come immagini (ricostruite grazie agli input provenienti dal nervo ottico). Nel caso in cui le cellule della retina siano danneggiate la visione viene meno. Nella retina esistono circa 131 milioni di cellule sensibili alla luce (fotorecettori). Se ne distinguono due tipi:
- I coni, così chiamati per la loro forma, recepiscono soprattutto i particolari delle immagini e i vari colori; ve ne sono di tre tipi diversi, che riconoscono rispettivamente il rosso, il verde e il blu (una loro combinazione viene però interpretata dal cervello come un colore specifico). Nella retina ve ne sono circa 6 milioni e si concentrano soprattutto al centro (macula); quindi, sono ovviamente fondamentali per la visione centrale (lettura, riconoscimento dei volti, guida, ecc.). La loro capacità di distinguere i dettagli è circa cento volte superiore rispetto ai bastoncelli.

- I bastoncelli, dalla linea allungata ed affusolata, reagiscono prevalentemente al contrasto fra il chiaro e lo scuro nonché al movimento degli oggetti. Sono circa 125 milioni e si concentrano nella zona periferica della retina, dove sono presenti in misura venti volte superiore rispetto ai coni. Nella parte centrale della nostra retina prevalgono dunque i coni, mentre sono pochi i bastoncelli. Questi ultimi nell’area periferica sono, invece, assai più numerosi; grazie ad essi possiamo avvertire un corpo in movimento (con la coda dell’occhio), anche se non siamo in grado di distinguerne i particolari (perché il riconoscimento fine avviene per mezzo dei coni).

Quante persone colpisce la retinite pigmentosa?
La retinite pigmentosa si stima che colpisca 1-5 persone su 10000 [1] a livello genetico. Complessivamente le degenerazioni retiniche ereditarie colpirebbero circa 2 milioni di persone nel mondo [2], di cui circa 1,5 milioni sarebbero affette da retinite pigmentosa. Purtroppo attualmente non esiste ancora alcuna cura efficace, nonostante diverse équipe di ricerca ci stiano lavorando.
Molto spesso la retinite pigmentosa compare tra la pubertà e l’età matura, ma non sono rari gli esempi di bambini colpiti nella prima infanzia. La capacità visiva della persona colpita subisce una riduzione progressiva. L’unica informazione certa di cui gli scienziati dispongono è l’origine genetica della retinite pigmentosa, la quale viene trasmessa ereditariamente, di generazione in generazione, seguendo meccanismi ormai noti (i geni responsabili sarebbero almeno 64 [3]). Secondo altre fonti si supererebbero persino i 70 geni “difettosi”. [4]
Si possono fare figli con un malato di retinite pigmentosa?
Se si vogliono fare figli con una persona colpita da retinite pigmentosa (RP) è senz’altro opportuno prima ricorrere ad approfonditi esami genetici per poter quantificare i rischi di trasmissione della malattia.
Quali forme esistono di retinite pigmentosa?
Le forme genetiche di retinite pigmentosa sono essenzialmente quattro:
- autosomica dominante
a) può colpire maschi e femmine con pari frequenza;
b) non salta le generazioni. - autosomica recessiva
a) la malattia colpisce con pari frequenza entrambi i sessi;
b) la malattia salta le generazioni, anzi non è infrequente l’evenienza che in una famiglia coinvolta compaia a memoria d’uomo un solo caso, ovvero che sia simulata una forma sporadica. - legata al cromosoma X (cioè legata al sesso)
Secondo questo tipo di trasmissione ereditaria, risultano colpiti dalla malattia solo soggetti di sesso maschile, i quali però ereditano il gene patologico dalla madre che è portatrice sana; data una donna in tale condizione, il rischio di malattia per ogni figlio maschio è del 50%. - sporadica
Le forme sporadiche (circa il 30% di tutti i casi) prevedono la presenza di un unico caso a memoria d’uomo in una famiglia. La sporadicità è solo una constatazione familiare, ma è molto difficile escludere l’eredità recessiva oppure quella legata al sesso, se la persona affetta è un maschio.
Bisogna differenziare, inoltre, la retinite pigmentosa primaria, in cui esiste solo l’interessamento oculare, da quella associata a malattie extraoculari.
Quali sono i sintomi?
I principali sintomi che possono indurre il medico a sospettare di trovarsi di fronte ad un caso di retinite pigmentosa sono essenzialmente due:
- Cecità crepuscolare e notturna
Ci può essere difficoltà a vedere in condizioni di scarsa illuminazione (muoversi e guidare di sera o di notte) o un ritardato adattamento quanto si passa da ambienti illuminati a quelli oscuri. Questo fenomeno è dovuto al fatto che, almeno per la maggior parte dei casi, la malattia – almeno nelle prime fasi del suo sviluppo – aggredisce prevalentemente i bastoncelli. - Restringimento del Campo Visivo (visione tubulare)
Si manifesta con una difficoltà a percepire oggetti posti lateralmente oppure non si notano né gradini né ostacoli bassi. L’alterazione del campo visivo è progressiva e può coinvolgere anche la parte centrale della retina, con perdita delle capacità visive centrali. La velocità di progressione della malattia e l’età di comparsa dei sintomi variano in relazione a molti fattori tra cui il modello di trasmissione genetica. Si riscontra, inoltre, un’aumentata sensibilità all’abbagliamento (che si verifica anche con molte altre patologie oculari); svaniscono i contrasti e diventa difficile percepire l’ambiente circostante.

Come si effettua la diagnosi?
La diagnosi di retinite pigmentosa in presenza di tutti i sintomi classici è facile ed è di pertinenza dell’oculista (e del genetista). Vengono generalmente effettuati:
- l’esame del visus;
- l’esame del fondo dell’occhio e la sua fotografia;
- l’esame del campo visivo;
- l’elettroretinogramma;
- la fluorangiografia.
La malattia può essere diagnosticata fin dall’infanzia, nell’adolescenza e, non di rado, anche in età adulta. Nei casi dubbi, la diagnosi si basa su tutti i dati clinici ottenibili (età di esordio, modalità evolutive, eventuale associazione con altri sintomi oculari od a carico di altri organi ed apparati) e su un approfondito studio elettrofisiologico (elettroretinogramma ed elettroculogramma) ed adattometrico. Possono risultare utili e complementari lo studio del senso cromatico e la fluoroangiografia retinica. E’ necessario, inoltre, esaminare tutto il nucleo familiare, allo scopo di definire il tipo di trasmissione ereditaria.
L’esame del visus
Permette di valutare l’acutezza visiva nella porzione centrale della retina. Consiste nella lettura di caratteri di varia grandezza alla distanza di 5 metri.
L’esame del fondo oculare
Ha lo scopo di valutare la morfologia della retina e di ricercare la presenza di caratteristiche macchie di pigmento sulla superficie retinica, che nella malattia hanno un aspetto tipico detto ad osteoblasti. Talune forme di retinite, pur presentando gli stessi sintomi, non sono però caratterizzate dalla presenza di macchie sul fondo dell’occhio.
L’esame del campo visivo
Permette di valutare la sensibilità retinica ad uno stimolo luminoso nelle varie zone della retina. È utile per avere una documentazione oggettiva delle difficoltà percepite dal paziente durante il movimento per monitorare l’evoluzione della malattia.
L’elettroretinogramma (ERG)
Consiste nella registrazione dell’attività elettrica della retina in risposta a particolari stimoli luminosi. Permette di valutare in modo distinto la funzionalità dei due tipi di fotorecettori (i coni ed i bastoncelli). L’elettroretinogramma è un esame molto importante per diagnosticare la retinite pigmentosa, poichè anche quando la malattia è ancora in fase iniziale, il tracciato che ne deriva è quasi sempre estinto e molto appiattito.
La fluorangiografia (FAG)
Si inietta per via endovenosa una sostanza fluorescente e si eseguono delle fotografie della retina in tempi diversi. Infatti, tramite la circolazione sanguigna, la sostanza fluorescente giunge sino alla retina, rendendo visibili, colorandole, le arterie, i capillari e le vene ed indica lo stato delle loro pareti.
Che decorso ha la malattia?
Il decorso della malattia ha una durata estremamente variabile, ma comunque è sempre progressivo ed invalidante. Nella maggioranza dei casi i sintomi si aggravano e purtroppo il campo visivo si restringe sempre più fino a chiudersi completamente. Compaiono inoltre anche altri disturbi, come l’abbagliamento, l’incapacità di distinguere i colori e una particolare forma di cataratta. Malauguratamente l’esito finale è, in molti casi, la cecità assoluta.
Si può curare?

Attualmente non è considerata una malattia curabile. Molte speranze erano state riposte sulle possibilità della terapia iperbarica (ossigenoterapia; OTI) per arrestare l’evoluzione della malattia. Numerosi studi hanno effettivamente registrato una risposta cellulare positiva, dimostrata anche strumentalmente con l’elettroretinogramma (ERG), che evidenziava un incremento della sua ampiezza statisticamente significativo nei pazienti trattati in camera iperbarica con ossigeno. Purtroppo però l’ossigenoterapia non è risolutiva e non elimina il problema all’origine.
Sono molte le strade di ricerca aperte, nonostante pochi progressi concreti siano stati compiuti fino ad oggi sul fronte delle cure clinicamente possibili. Attualmente i filoni più promettenti sono la terapia genica, il ricorso a cellule staminali e il trapianto di retina o di fotorecettori.
Secondo uno studio pubblicato il 16 novembre 2016 su Nature [5] da ricercatori che lavorano in California, all’orizzonte ci sarebbero nuove possibilità di trattamento grazie a una nuova tecnica pionieristica di correzione genetica (approfondisci). Infatti, in seguito a una sperimentazione condotta su cavie animali in cui era stata indotta artificialmente la retinite pigmentosa, si è avuto un parziale recupero della loro vista. Tuttavia, al momento in cui scriviamo questo risultato non è ancora stato esteso agli esseri umani [6]. Lo stesso discorso vale per uno studio successivo di Ophthalmology [7], condotto sempre con tecnica CRISP di editing genetico da ricercatori della Columbia University (Usa) su cavie murine.
Qualche risultato positivo sembrerebbe, inoltre, avere avuto l’impiego del fattore di crescita nervoso (NGF) sotto forma di collirio sperimentale. Tuttavia i casi che, al momento in cui scriviamo, avrebbero tratto giovamento dalla sua instillazione risulterebbero essere un’esigua minoranza [8].
Infine uno studio pubblicato il 29 settembre 2017 sulla rivista Stem Cell Research & Therapy [9] ha fornito risultati sperimentali limitati ma, per certi versi, incoraggianti: è stata parzialmente rigenerata la retina in otto pazienti impiegando staminali embrionali (il cui impiego è però vietato in Italia e in molti altri Paesi), con un monitoraggio durato due anni e risultati non omogenei (miglioramenti del visus nella maggior parte degli occhi, acuità visiva stabile o peggiorata dopo 24 mesi negli altri). Eppure, “il nostro studio – scrivono ricercatori cinesi del Southwest Eye Hospital, Third Military Medical University – ha confermato per la prima volta, in pazienti divenuti ciechi a causa della retinite pigmentosa, la sicurezza nel lungo periodo e la fattibilità del ripristino della vista mediante terapia con cellule staminali”.
Bisognerà però attendere ulteriori ricerche per capire meglio la portata effettiva di tutte queste sperimentazioni.
Le vitamine possono aiutare?
Secondo alcuni studi l’assunzione di elevati dosaggi di vitamina A potrebbe contribuire a rallentare la progressione della malattia, mentre la vitamina E avrebbe un effetto contrario.
In un esperimento condotto nel lontano 1993 si fa presente che i malati (18-49 anni), nonostante l’assunzione massiccia della vitamina A, subivano comunque un calo della vista (mediamente perdevano una riga l’anno nell’ottotipo). Una recente ricerca condotta su studi precendenti (metastudio), pubblicata a dicembre del 2013, ha concluso comunque che non esistono prove sufficienti che dimostrino l’efficacia della vitamina A (né dell’olio di pesce) nel rallentare la progressione della retinite pigmentosa.
Terapia genica
Si propone di identificare i geni responsabili della malattia, per poter poi intervenire con le tecniche più sofisticate dell’ingegneria genetica. In particolare si mira alla sostituzione dei geni “difettosi” con geni sani e, nel caso della trasmissione autosomica dominante, alla disattivazione dei geni nocivi [10] . Per veicolare il materiale genetico ‘buono’ generalmente si usa un virus vettore (quello del raffreddore) preventivamente reso innocuo. Come se fosse un cavallo di Troia, il rinovirus è in grado di trasportare parti del dna necessarie alla ‘riparazione’ genetica: effettuando delle iniezioni sotto la retina si punta a curare la malattia. Tale approccio ha avuto in parte successo soprattutto sui bambini colpiti da un’altra malattia genetica che colpisce la retina: l’amaurosi congenita di Leber. Tuttavia la retinite pigmentosa coinvolge un numero superiore di geni e, dunque, è più difficile da trattare geneticamente.
Cellule staminali
Con l’impiego di cellule staminali si sta tentando di rimpiazzare i fotorecettori persi a causa della degenerazione retinica. Sono stati ottenuti risultati incoraggianti con staminali embrionali impiantate nella retina dei topi, ma solo in un quarto dei casi; in oltre la metà dei casi, invece, si sono avuti distacco di retina e sviluppo di tumori [11]. Tuttavia uno studio già citato, pubblicato nel 2017, ha assicurato che mediante una tecnica più recente non sono stati indotti tumori. Tuttavia non si possono mai escludere, in linea di principio, complicanze.
Dunque, bisognerà attendere molte altre ricerche affinché l’impiego delle staminali possa essere perfezionato sugli esseri umani, ad esempio nei Paesi dove l’uso delle cellule embrionarie è legale. In alternativa si potrà presumibilmente ricorrere alle cellule staminali riprogrammate (ottenute da cellule adulte), “ringiovanite” mediante tecniche di ingegneria genetica; ma occorreranno ancora molti altri studi affinché le staminali possano essere usare proficuamente nell’uomo a fini di rigenerazione retinica.
Tuttavia è bene chiarire che attualmente non esiste al mondo nessun protocollo clinico approvato che preveda l’uso di staminali per trattare efficacemente la retinite pigmentosa (né altre malattie retiniche degenerative). [12]
Trapianti
L’intento è quello di mettere a punto una tecnica che renda possibile il trapianto di tessuto retinico o, per lo meno, l’innesto di cellule sane su retine malate. Attualmente, tuttavia, il trapianto di retina non è mai stato effettuato con successo (né del tutto né in parte).
Immunologia
Si prefigge di verificare alcune teorie che ipotizzerebbero un’alterazione del sistema immunitario che potrebbe essere il principale fattore scatenante della malattia.
Il futuro dei trattamenti
Il futuro delle cure potrebbe risiedere, nel caso della retinite pigmentosa, nelle cellule staminali e nella terapia genica. In alcuni malati che hanno perso la vista a causa di qtia sono state impiantate sperimentalmente retine elettroniche, restituendo loro (quando l’intervento ha avuto successo) una visione molto limitata.
Vedi anche: Rayapudi S, Schwartz SG, Wang X, Chavis P., “Vitamin A and fish oils for retinitis pigmentosa“, Cochrane Database Syst Rev. 2013 Dec 19;12:CD008428. doi: 10.1002/14651858.CD008428.pub2.
[2] Attualmente si ritiene che coinvolgano circa 250 geni (con 4500 mutazioni), almeno secondo lo studio scientifico di Lobanova ES, Finkelstein S, Li J, Travis AM, Hao Y, Klingeborn M, Skiba NP, Deshaies RJ, Arshavsky VY, “Increased proteasomal activity supports photoreceptor survival in inherited retinal degeneration“, Nat Commun. 2018 Apr 30;9(1):1738. doi: 10.1038/s41467-018-04117-8
[3] Zhang L, Du J, Justus S, Hsu CW, Bonet-Ponce L, Wu WH, Tsai YT, Wu WP, Jia Y, Duong JK, Mahajan VB, Lin CS, Wang S, Hurley JB, Tsang SH, “Reprogramming metabolism by targeting sirtuin 6 attenuates retinal degeneration“, J Clin Invest. 2016 Nov 14. pii: 86905. doi: 10.1172/JCI86905
[5] Juan Carlos Izpisua Belmonte et al., “In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration“, Nature, 2016 Nov 16, doi: 10.1038/nature20565 (Epub ahead of print)
[6] Si veda: “New gene-editing technology partially restores vision in blind animals“, Salk News, 16 novembre 2016
[7] Tsai, Yi-Ting et al., “Clustered Regularly Interspaced Short Palindromic Repeats-Based Genome Surgery for the Treatment of Autosomal Dominant Retinitis Pigmentosa, Ophthalmology, 2018
[9] Yong Liu et al., “Long-term safety of human retinal progenitor cell transplantation in retinitis pigmentosa patients“, Stem Cell Research & Therapy ,2017
[10] Un interruttore generale per combattere la retinite pigmentosa, Le Scienze, 26 gennaio 2011
[11] “Transplantation of Reprogrammed Embryonic Stem Cells Improves Visual Function in a Mouse Model for Retinitis Pigmentosa”, by Wang, Nan-Kai; Tosi, Joaquin; Kasanuki, Jennifer Mie; Chou, Chai Lin; Kong, Jian; Parmalee, Nancy; Wert, Katherine J.; Allikmets, Rando; Lai, Chi-Chun; Chien, Chung-Liang; Nagasaki, Takayuki; Lin, Chyuan-Sheng; Tsang, Stephen H., Transplantation, 15 February 2010, doi: 10.1097/TP.0b013e3181d45a61
[12] L’unico impiego approvato a livello oculare è quello delle staminali corneali
Scheda informativa a cura dell’Agenzia internazionale per la prevenzione della cecità-IAPB Italia onlus
Leggi le condizioni generali di consultazione di questo sito
Pagina pubblicata il 3 aprile 2009. Ultimo aggiornamento: 19 giugno 2019.
Ultima revisione scientifica: 16 marzo 2018.[6101
