



 Ecco la casa
Ecco la casa
‘parlanté per ciechi ed ipovedenti Esistono ormai numerosi apparecchi dotati di sintetizzatore vocale
23 novembre 2007 – La casa ‘parlanté non appartiene più solo al
regno delle fiabe. Sono oggi già disponibili una serie di apparecchi con sintetizzatore
incorporato che dà preziose informazioni verbali ai ciechi ed agli ipovedenti.
Dal riconoscitore di colori agli orologi, dal metro avvolgibile agli strumenti
utili per controllare il proprio stato di salute (ad esempio il termometro, il
misuratore di pressione e di glicemia): sono molti gli apparecchi che possono
parlare grazie alle nuove tecnologie, così come avviene nel mondo dell’infanzia.
Ci sono strumenti molto utili,
tra cui il contagocce elettronico, ma anche il riconoscitore di colori non è da meno:
i modelli più sofisticati riescono persino a valutare la sfumatura, magari distinguendo
modelli più sofisticati riescono persino a valutare la sfumatura, magari distinguendo
un rosa shocking da uno antico o un rosso carminio da uno vermiglio.
Inoltre, esistono attrezzi come il bastone ‘intelligenté, detto ‘intercetta-ostacoli’: funziona grazie
all’emissione continua di onde, suonando quando si è in
presenza di un ostacolo. Senza parlare della vernice magnetica, che permette al
non vedente di seguire un percorso prestabilito: è stata sperimentata con successo
negli Stati Uniti e di recente è arrivata anche in Italia.
Le nuove tecnologie sono di
grande aiuto per gli ipovedenti e i non vedenti: basti pensare ai cellulari con
sintesi vocale e ai software di screen-reading,
che leggono automaticamente i contenuti dello schermo. Inoltre, tra i giovani
disabili visivi è ormai in voga la
MP-3 mania: registrano spesso conversazioni ed informazioni
utili col lettore di file audio.
![]() Servizio televisivo su Apri gli Occhi!TG-R Rai-LiguriaOre 19 del 21-11-2007
Servizio televisivo su Apri gli Occhi!TG-R Rai-LiguriaOre 19 del 21-11-2007


![]() Servizio televisivo su Apri gli Occhi!TG-R Rai-VenetoOre 14 del 13-11-2007
Servizio televisivo su Apri gli Occhi!TG-R Rai-VenetoOre 14 del 13-11-2007
![]() Servizio televisivo su Apri gli
Servizio televisivo su Apri gli
Occhi!TG-R Rai-FriuliOre 19 del 17-11-2007
L’atrofia girata è una distrofia corioretinica ereditaria: colpisce la coroide e la retina danneggiandoli irreparabilmente. Viene trasmessa in forma autosomica recessiva (entrambi i genitori presentano il difetto genetico). In genere i sintomi si avvertono attorno ai 20-30 anni.
Da una mutazione di un gene responsabile della produzione dell’enzima ornitina aminotransferasi, coinvolto nel metabolismo dell’ornitina (proteina che viene sintetizzata anche attraverso la dieta). Il danno oculare si manifesta con aree di atrofia retinica: si perde progressivamente la funzionalità della retina.
I sintomi consistono soprattutto in disturbi della visione notturna e nella riduzione periferica del campo visivo. La visione centrale viene, invece, conservata anche in età molto avanzata. La progressione della malattia è, comunque, molto lenta.
La diagnosi viene effettuata con l’esame del fondo oculare: si evidenziano aree focali in periferia (e media periferia) di atrofia corioretinica. La fluorangiografia non è esame fondamentale per la diagnosi. L’elettro-oculogramma (EOG) si presenta alterato così come l’elettroretinogramma (ERG). È molto importante valutare il dosaggio dell’ornitina sierica, nelle urine ed eventualmente nel fluido cerebrospinale; infatti la sua presenza in quantità elevate si associa alla malattia.
Sono state riscontrate alterazioni della funzionalità epatica; in alcuni casi si presentano patologie della struttura delle ossa e anomalie a livello di elettroencefalogramma.
Alcune ricerche hanno evidenziato che, diminuendo l’apporto di arginina attraverso la dieta, si riducono i livelli sierici di ornitina e, quindi, si rallenta la progressione della malattia.
La malattia di Best è una patologia ereditaria della retina. Viene trasmessa in forma autosomica dominante (un genitore trasmette il difetto genetico al figlio o alla figlia).
La malattia è causata da una mutazione di un gene (chiamato VMD2, localizzato sul cromosoma 11q13), che nella retina regola il trasporto di determinate sostanze (acidi grassi polinsaturi) e comporta l’accumulo di un materiale di scarto biologico (lipofuscina) in uno strato chiamato epitelio pigmentato retinico.
I sintomi consistono soprattutto nella riduzione della vista generalmente in forma lieve, con una progressione lenta. I pazienti riferiscono disturbi maggiori nelle visione da vicino, a cui si possono accompagnare la distorsione dell’immagine e gli scotomi centrali (macchie nere nel campo visivo).
La diagnosi viene effettuata con l’esame del fondo oculare: si possono osservare caratteristiche alterazioni della macula (zona centrale della retina) che compaiono già dall’infanzia e adolescenza, con la presenza di una lesione tipica (detta vitelliforme) che, col passare degli anni, può evolvere in altri stadi associandosi alla riduzione della vista; la fluorangiografia può essere di aiuto nella diagnosi. Tuttavia, l’esame fondamentale per accertare la malattia è l’elettroculogramma (EOG) che si presenta alterato in associazione ad un elettroretinogramma (ERG) normale.
No, non sono stati descritti casi di altre malattie associati alla malattia di Best.
Al momento non ci sono terapie efficaci nel bloccare la progressione del danno o per poter guarire dal danno precedente. Nei casi in cui l’evoluzione della lesione maculare determina l’insorgenza di neovascolarizzazioni si possono eseguire dei trattamenti laser per il controllo delle stesse. Può essere importante l’uso di ausili per ipovedenti che permettono di sfruttare al meglio il residuo visivo.

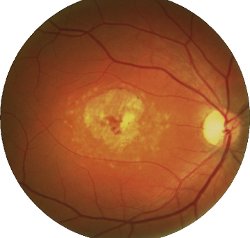
La malattia (o maculopatia) di Stargardt è una patologia ereditaria della retina che si manifesta generalmente prima dei vent’anni. Il più delle volte viene trasmessa in forma autosomica recessiva (entrambi i genitori presentano il difetto genetico [[pur potendo essere portatori sani]]), ma sono stati descritti anche casi di forme autosomiche dominanti (un solo genitore trasmette il difetto del DNA).
La malattia è provocata da una mutazione di un gene (ABCA4), che comporta l’accumulo di materiale di scarto (simile alla lipofuscina) nella retina (in uno strato esterno chiamato epitelio pigmentato). Questo materiale è originato dalla degradazione di sostanze presenti nei coni e nei bastoncelli (fotorecettori retinici).
Consistono soprattutto nella riduzione della visione centrale (spesso in forma grave) che può iniziare durante l’adolescenza o anche nell’infanzia. Inoltre, chi ne è affetto può lamentare disturbi nella percezione dei colori (discromatopsia), scotomi centrali (macchie nere nel campo visivo) e fotofobia (intolleranza alla luce).
La diagnosi viene effettuata con l’esame del fondo oculare: si possono osservare caratteristiche alterazioni della macula (zona centrale della retina); tuttavia ciò deve essere confermato dalla fluorangiografia. Al contrario, gli esami elettrofunzionali (ERG, EOG, PEV) non sono in questo caso importanti per la diagnosi, in quanto non presentano alterazioni caratteristiche.
Indicativamente una persona su diecimila. Se entrambi i genitori sono portatori sani della patologia la probabilità che un figlio si ammali è del 25% (una probabilità su quattro).
No, non sono stati descritti casi di altre malattie sistemiche associate.
Al momento non ci sono terapie efficaci nel bloccare la progressione della malattia o per poter guarire dal danno già esistente (alcuni scienziati stanno però tentando di usare sperimentalmente cellule staminali per rallentare la degenerazione retinica). Tuttavia, è di grande aiuto l’utilizzo di ausili per ipovedenti che permettono di sfruttare al meglio ciò che resta della vista (residuo visivo). Anche la riabilitazione visiva consente di dare risultati soddisfacenti, con eventuali sedute di fotostimolazione e un supporto psicologico.
![]() INTERVISTA ALL’AVV. GIUSEPPE CASTRONOVO
INTERVISTA ALL’AVV. GIUSEPPE CASTRONOVO
Presidente della IAPB Italia
Agenzia internazionale per la prevenzione della cecità su “Apri gli Occhi!” target=”_blank” 2007-8